Abstract
Electrochemically generated amidyl radical species produced distinct inter- or intramolecular hydroamination reaction products via a proton-coupled electron transfer (PCET) mechanism. Cyclic voltammetry (CV) analysis indicated that the chemoselectivity was derived from the size of the hydrogen bond complex, which consisted of the carbamate substrate and phosphate base, and could be controlled using 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as an additive. These results provide fundamental insights for the design of PCET-based redox reaction systems under electrochemical conditions.
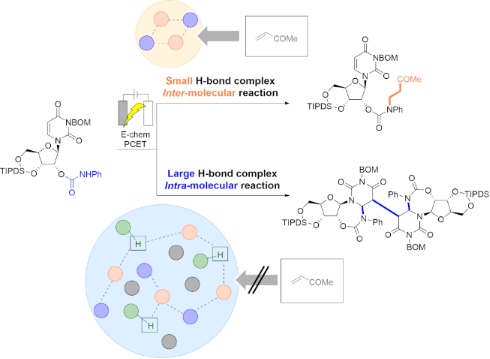
Graphical Abstract
Introduction
Proton-coupled electron transfer (PCET) enables the generation of various radical species under ambient conditions (Figure 1, top) [1]. In PCET processes, hydrogen bond formation between weak bases and acidic X–H bonds (X = N, O, C) is a key step, which is followed by concerted proton- and electron-transfer to give the corresponding radical species through oxidative X–H bond cleavage. One such species is the amidyl radical, which is broadly synthetically useful as a nitrogen source in hydroamination reactions and as a hydrogen atom transfer (HAT) reagent for remote C–H activation [2-8]. Recent advances in photoredox and electrochemical PCET reactions have significantly expanded the substrate scope of amidyl-radical-based molecular transformations because the harsh acidic and high-temperature conditions required in the classical Hofmann–Löffler–Freytag reaction can be avoided [9].
![[1860-5397-20-27-1]](/bjoc/content/figures/1860-5397-20-27-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Application of amidyl radical species generated by PCET.
Figure 1: Application of amidyl radical species generated by PCET.
The initial aim of this study was the electrochemical generation of an amidyl radical as a HAT source for the synthesis of 1’-C functionalized nucleosides via the generation of an anomeric radical species from uridine derivative 1 (Figure 1, bottom) [10]. Although the HAT reaction failed, remarkable inter- and intramolecular chemoselectivities were observed in the hydroamination reaction. We investigated this phenomenon and found that complete inter-/intramolecular chemoselectivity could be achieved by modifying the reaction conditions, despite the presence of both inter- and intramolecular radical acceptor moieties. Therefore, we investigated the origin of this selectivity under electrochemical conditions.
Results and Discussion
Anodic oxidation of uridine derivative 1 was performed in a CH2Cl2/Bu4NPF6 (0.1 M) electrolyte system using a carbon felt (CF) anode and a Pt cathode in the presence of methyl vinyl ketone (MVK) as a radical acceptor (Table 1). Tetrabutylammonium dibutyl phosphate (phosphate base), which operates as a PCET initiator through hydrogen bond formation with the N–H bond of amide/carbamate [11], was used as an additive. As a result, N-alkylated product 3 was exclusively obtained, implying that the expected HAT at the 1’-C position to afford 2 (Table 1, entry 1) had not occurred. In contrast, the reaction efficiency was significantly decreased in the absence of the phosphate base (Table 1, entry 2), and electricity is necessary to proceed the reaction (Table 1, entry 3); thus, the phosphate base plays a crucial role in N-alkylation, while its basicity is insufficient to promote aza-Michael addition (pKa of the conjugate acid of the phosphate base is 1.72 in H2O) [12]. Furthermore, N-alkylation proceeded in a divided cell (anodic chamber); thus, the possibility of conjugate addition of a cathodically generated carbamate anion was ruled out, prompting us to consider that N-alkylation proceeded via a radical mechanism. On the other hand, the addition of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) led to the predominant formation of cyclized dimer 4 without N-alkylation, whereas the use of AcOH provided N-alkylated product 3 (Table 1, entries 5 and 6). When acetonitrile (MeCN) was used as the solvent, cyclized dimer 4 was obtained (Table 1, entry 7).
Table 1: Electrochemical oxidation of 1 under varying conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-27-i1.svg?max-width=637&scale=1.0)
|
|||
| Entry | Deviation from standard conditions | Yield [%]a | Recovered 1 [%]a |
| 1 | none | 57, 49b (3) | 17 |
| 2 | without phosphate base | 13 (3) | 76 |
| 3 | without electricity | N.R. | 92 |
| 4 | divided cell (anodic chamber) | 41 (3) | 27 |
| 5 | HFIP (2 equiv) as an additive | 42, 27b (4) | 32 |
| 6 | AcOH (2 equiv) as an additive | 10 (3) | 51 |
| 7 | MeCN instead of CH2Cl2 | 17 (4) | 28 |
aYield was determined based on 1H NMR by using benzaldehyde as an internal standard, and recovery rate of 1 was determined by the integral of H-1’ proton. bIsolated yield.
Next, 1 was subjected to cyclic voltammetry (CV) measurements under varying conditions (Figure 2). An oxidation wave was observed at approximately +1.4 V (Figure 2A). The oxidation current of this wave decreased significantly in the presence of a phosphate base and the subsequent addition of HFIP enhanced this phenomenon (Figure 2B, grey line). In contrast, using AcOH instead of HFIP did not affect the oxidation current (Figure 2B, blue line). We considered that the inter- and intramolecular chemoselectivities were derived from the pKa of the proton sources.
![[1860-5397-20-27-2]](/bjoc/content/figures/1860-5397-20-27-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (A) Effect of phosphate base on the cyclic voltammogram of 1. (B) Cyclic voltammograms of 1 in the presence of additives (AcOH or HFIP). (C) Comparison of oxidation potentials of 1 using Bu4NOAc or Bu4NCl. (D) Cyclic voltammograms for the cathodic side. All cyclic voltammograms were recorded in CH2Cl2/Bu4NPF6 (0.1 M). Sample concentration was 0.01 M. A glassy carbon anode (φ 3 mm) and Pt cathode (φ 3 mm) were used. Scan rate = 100 mV/s.
Figure 2: (A) Effect of phosphate base on the cyclic voltammogram of 1. (B) Cyclic voltammograms of 1 in the ...
The pre-organization of the amide substrate and phosphate bases is an important process in PCET [13]. Recently, Gschwind et al. published a detailed NMR spectroscopic analysis of a PCET-mediated hydroamination reaction, which indicated that the pKa of the proton source (PhSH or PhOH in the study) determines the size of the hydrogen bond complex. PhSH as the more acidic additive (pKa = 6.62 in H2O) provided better results in the PCET-induced intramolecular hydroamination reaction compared to the less acidic PhOH (pKa = 9.95 in H2O) because PhSH supplied free protons (H+) and contributed to the persistence of small aggregates composed of the amide and phosphate base [14]. On the other hand, owing to the insufficient dissociation constant between the proton and phenoxide in PhOH, the PhOH molecule is included in the hydrogen bond network along with the tetrabutylammonium cation (Bu4N+) to form a large aggregate. The hydrogen bonding between the amide and phosphate base in the small aggregates was stronger than in the large aggregates, which significantly enhanced amidyl radical generation through the PCET mechanism.
The above studies provided us with valuable insights into the intriguing electrochemical behavior of 1 (Figure 3). Hydrogen bond formation between 1 and the phosphate base yielded small aggregates, the interaction efficiency of which with the electrode surface was lower than that of 1 because the relatively large hydrodynamic radius of the aggregates decreased the number of electrode-accessible molecules. This increase in the hydrodynamic radius resulted in a decrease in the oxidation current. In the present study, HFIP (pKa = 9.30 in H2O) [15] is less acidic than AcOH (pKa = 4.76 in H2O) with a pKa value similar to that of PhOH, which forms large aggregates under PCET conditions, as described above. Therefore, analogously, HFIP is expected to be included in the hydrogen-bonded complex. The resulting large aggregates further impeded access to the electrode surface, and a further decrease in the oxidation current was observed in the presence of HFIP (Figure 2B, grey line). In contrast, the more acidic AcOH supplied free protons, which enabled the persistence of small aggregates; thus, the current was not affected by the presence of AcOH (Figure 2B, blue line). However, in the presence of AcOH, the N-alkylation yield was low (Table 1, entry 6) owing to the competitive Kolbe oxidation of the cathodically generated acetate anion. In fact, the oxidation potential of Bu4NOAc is lower than that of 1 (Figure 2C, orange line).
![[1860-5397-20-27-3]](/bjoc/content/figures/1860-5397-20-27-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Plausible models illustrating the size effect of the hydrogen bond complex on the interaction efficiency with electrode surfaces.
Figure 3: Plausible models illustrating the size effect of the hydrogen bond complex on the interaction effic...
A decrease in the oxidation current can be considered as a decrease in the diffusion coefficient of the hydrogen bond complex; thus, we attempted to reproduce the CV pattern by computational simulation (Figures S1 and S2 in Supporting Information File 1) [16]. The results indicated that an excessively small diffusion coefficient (1/10- or 1/100-fold) is required to reproduce a CV pattern similar to that observed experimentally. Because the reported diffusion coefficient is only twice as small as that of the sole amide molecule [14], this simulated value is unrealistic, and we assumed that the diffusion coefficient did not affect the oxidation current.
In cathodic events, the reduction of CH2Cl2 primally occurred under standard conditions because the reduction wave of the blank solution appeared at approximately −1.0 V (Figure 2D, blue line). The resulting cathodically generated chloride ion (Cl−) has a lower oxidation potential than 1 (Figure 2C, grey line); thus, it was subsequently oxidized on the anode to afford the halonium ion (Cl+), which can react with 1 to form unstable N−Cl species (B) in situ (Figure 4). Although we cannot detect the chlorinated intermediate of 1, electrolysis of N-propylcarbamate derivative under standard conditions gave the corresponding N−Cl species (C) as an unstable compound. We considered that this result as direct evidence for the plausibility of the existence of N−Cl species which driving the minor reaction pathway.
![[1860-5397-20-27-4]](/bjoc/content/figures/1860-5397-20-27-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Plausible mechanism for the inter-/intramolecular hydroamination of 1.
Figure 4: Plausible mechanism for the inter-/intramolecular hydroamination of 1.
Further single-electron reduction affords the amidyl radical [17], which can react with MVK. Because N-alkylation also proceeded in the absence of a phosphate base but in a low yield (Table 1, entry 2), it can be concluded that only the N−Cl species contributed to N-alkylation in this case.
Based on the experimental and simulation results, we propose a plausible mechanism for the inter- and intramolecular hydroamination of 1 (Figure 4). In the N-alkylation reaction, anodic oxidation of a small hydrogen-bonded complex produces amidyl radical A. The hydrophobic MVK molecule was excluded from the highly polar environment of this complex, but the resulting amidyl radical could access MVK because it still had a large surface area for interaction with the solution interface. As mentioned above, the amidyl radical can also be generated through N−Cl species B.
However, the large hydrogen-bond complex, which included HFIP, prevented amidyl radical access to MVK. In this case, intramolecular radical trapping by the uracil nucleobase was preferred, leading to the formation of the cyclized alkyl radical D. Continuous radical recombination furnished dimer 4.
Conclusion
We observed additive-controlled inter- and intramolecular chemoselectivity in the hydroamination of 1. Detailed CV analysis indicated that the size of the hydrogen bond complex determined the selectivity, and HFIP played a crucial role in expanding the hydrogen bond network. These results provide fundamental insights beneficial for the design of PCET-based redox reaction systems under electrochemical conditions.
Experimental
General procedure of anodic oxidation
Compound 1 (145 mg, 0.2 mmol), Bu4NPF6 (387 mg, 1 mmol), CH2Cl2 (10 mL), phosphate base (90 mg, 0.2 mmol) and methyl vinyl ketone (32.7 μL, 0.4 mmol) were added to a test tube, which was then subjected to a constant electrical current of 5 mA (3 F/mol, 57.9 C) through the CF anode (1 × 1 cm) and the Pt cathode (1 × 1 cm). The reaction mixture was concentrated in vacuo and Et2O (20 mL) was added. The resulting precipitate was removed by filtration through a short silica gel pad under reduced pressure. The filtrate was concentrated in vacuo and the resulting residue was subjected to 1H NMR spectroscopy or column chromatography. A divided-cell experiment was performed using an H-type cell (4G glass filter). Compound 1 (0.2 mmol), Bu4NPF6 (387 mg, 1 mmol), phosphate base (90 mg, 0.2 mmol), CH2Cl2 (10 mL), and methyl vinyl ketone (32.7 μL, 0.4 mmol) were added to the anode chamber, and CH2Cl2 (10 mL), and Bu4NPF6 (387 mg, 1 mmol) were added to the cathode chamber. The anolyte was transferred to a round-bottomed flask, and the solvent was removed in vacuo. Et2O (20 mL) was added to the crude mixture, and the resulting precipitate was removed by filtration through a short silica gel pad under reduced pressure. The filtrate was concentrated in vacuo and the resulting residue was subjected to 1H NMR spectroscopy or column chromatography.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, CV simulation, copies of NMR spectra. | ||
| Format: PDF | Size: 1.3 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Murray, P. R. D.; Cox, J. H.; Chiappini, N. D.; Roos, C. B.; McLoughlin, E. A.; Hejna, B. G.; Nguyen, S. T.; Ripberger, H. H.; Ganley, J. M.; Tsui, E.; Shin, N. Y.; Koronkiewicz, B.; Qiu, G.; Knowles, R. R. Chem. Rev. 2022, 122, 2017–2291. doi:10.1021/acs.chemrev.1c00374
Return to citation in text: [1] -
Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339–3350. doi:10.1021/acs.accounts.9b00472
Return to citation in text: [1] -
Fazekas, T. J.; Alty, J. W.; Neidhart, E. K.; Miller, A. S.; Leibfarth, F. A.; Alexanian, E. J. Science 2022, 375, 545–550. doi:10.1126/science.abh4308
Return to citation in text: [1] -
Wang, F.; Stahl, S. S. Angew. Chem., Int. Ed. 2019, 58, 6385–6390. doi:10.1002/anie.201813960
Return to citation in text: [1] -
Zhu, Q.; Graff, D. E.; Knowles, R. R. J. Am. Chem. Soc. 2018, 140, 741–747. doi:10.1021/jacs.7b11144
Return to citation in text: [1] -
Davies, J.; Svejstrup, T. D.; Fernandez Reina, D.; Sheikh, N. S.; Leonori, D. J. Am. Chem. Soc. 2016, 138, 8092–8095. doi:10.1021/jacs.6b04920
Return to citation in text: [1] -
Choi, G. J.; Zhu, Q.; Miller, D. C.; Gu, C. J.; Knowles, R. R. Nature 2016, 539, 268–271. doi:10.1038/nature19811
Return to citation in text: [1] -
Xu, F.; Zhu, L.; Zhu, S.; Yan, X.; Xu, H.-C. Chem. – Eur. J. 2014, 20, 12740–12744. doi:10.1002/chem.201404078
Return to citation in text: [1] -
Hu, X.; Zhang, G.; Bu, F.; Nie, L.; Lei, A. ACS Catal. 2018, 8, 9370–9375. doi:10.1021/acscatal.8b02847
Return to citation in text: [1] -
Gimisis, T.; Chatgilialoglu, C. J. Org. Chem. 1996, 61, 1908–1909. doi:10.1021/jo952218n
Return to citation in text: [1] -
Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546–1556. doi:10.1021/acs.accounts.6b00272
Return to citation in text: [1] -
Kumler, W. D.; Eiler, J. J. J. Am. Chem. Soc. 1943, 65, 2355–2361. doi:10.1021/ja01252a028
Return to citation in text: [1] -
Darcy, J. W.; Koronkiewicz, B.; Parada, G. A.; Mayer, J. M. Acc. Chem. Res. 2018, 51, 2391–2399. doi:10.1021/acs.accounts.8b00319
Return to citation in text: [1] -
Berg, N.; Bergwinkl, S.; Nuernberger, P.; Horinek, D.; Gschwind, R. M. J. Am. Chem. Soc. 2021, 143, 724–735. doi:10.1021/jacs.0c08673
Return to citation in text: [1] [2] -
Dyatkin, B. L.; Mochalina, E. P.; Knunyants, I. L. Tetrahedron 1965, 21, 2991–2995. doi:10.1016/s0040-4020(01)96918-2
Return to citation in text: [1] -
Izumiya, R.; Atobe, M.; Shida, N. Electrochemistry 2023, 91, 112003. doi:10.5796/electrochemistry.23-67010
Return to citation in text: [1] -
Kim, H.; Kim, T.; Lee, D. G.; Roh, S. W.; Lee, C. Chem. Commun. 2014, 50, 9273–9276. doi:10.1039/c4cc03905j
Return to citation in text: [1]
| 1. | Murray, P. R. D.; Cox, J. H.; Chiappini, N. D.; Roos, C. B.; McLoughlin, E. A.; Hejna, B. G.; Nguyen, S. T.; Ripberger, H. H.; Ganley, J. M.; Tsui, E.; Shin, N. Y.; Koronkiewicz, B.; Qiu, G.; Knowles, R. R. Chem. Rev. 2022, 122, 2017–2291. doi:10.1021/acs.chemrev.1c00374 |
| 11. | Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546–1556. doi:10.1021/acs.accounts.6b00272 |
| 10. | Gimisis, T.; Chatgilialoglu, C. J. Org. Chem. 1996, 61, 1908–1909. doi:10.1021/jo952218n |
| 9. | Hu, X.; Zhang, G.; Bu, F.; Nie, L.; Lei, A. ACS Catal. 2018, 8, 9370–9375. doi:10.1021/acscatal.8b02847 |
| 2. | Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339–3350. doi:10.1021/acs.accounts.9b00472 |
| 3. | Fazekas, T. J.; Alty, J. W.; Neidhart, E. K.; Miller, A. S.; Leibfarth, F. A.; Alexanian, E. J. Science 2022, 375, 545–550. doi:10.1126/science.abh4308 |
| 4. | Wang, F.; Stahl, S. S. Angew. Chem., Int. Ed. 2019, 58, 6385–6390. doi:10.1002/anie.201813960 |
| 5. | Zhu, Q.; Graff, D. E.; Knowles, R. R. J. Am. Chem. Soc. 2018, 140, 741–747. doi:10.1021/jacs.7b11144 |
| 6. | Davies, J.; Svejstrup, T. D.; Fernandez Reina, D.; Sheikh, N. S.; Leonori, D. J. Am. Chem. Soc. 2016, 138, 8092–8095. doi:10.1021/jacs.6b04920 |
| 7. | Choi, G. J.; Zhu, Q.; Miller, D. C.; Gu, C. J.; Knowles, R. R. Nature 2016, 539, 268–271. doi:10.1038/nature19811 |
| 8. | Xu, F.; Zhu, L.; Zhu, S.; Yan, X.; Xu, H.-C. Chem. – Eur. J. 2014, 20, 12740–12744. doi:10.1002/chem.201404078 |
| 15. | Dyatkin, B. L.; Mochalina, E. P.; Knunyants, I. L. Tetrahedron 1965, 21, 2991–2995. doi:10.1016/s0040-4020(01)96918-2 |
| 14. | Berg, N.; Bergwinkl, S.; Nuernberger, P.; Horinek, D.; Gschwind, R. M. J. Am. Chem. Soc. 2021, 143, 724–735. doi:10.1021/jacs.0c08673 |
| 14. | Berg, N.; Bergwinkl, S.; Nuernberger, P.; Horinek, D.; Gschwind, R. M. J. Am. Chem. Soc. 2021, 143, 724–735. doi:10.1021/jacs.0c08673 |
| 17. | Kim, H.; Kim, T.; Lee, D. G.; Roh, S. W.; Lee, C. Chem. Commun. 2014, 50, 9273–9276. doi:10.1039/c4cc03905j |
| 13. | Darcy, J. W.; Koronkiewicz, B.; Parada, G. A.; Mayer, J. M. Acc. Chem. Res. 2018, 51, 2391–2399. doi:10.1021/acs.accounts.8b00319 |
| 12. | Kumler, W. D.; Eiler, J. J. J. Am. Chem. Soc. 1943, 65, 2355–2361. doi:10.1021/ja01252a028 |
| 16. | Izumiya, R.; Atobe, M.; Shida, N. Electrochemistry 2023, 91, 112003. doi:10.5796/electrochemistry.23-67010 |
© 2024 Okamoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.