Abstract
A substituent-dependent construction of novel A3B-porphyrins along with A4B2-hexaphyrins was realized by the reactions of N-tosylimines and meso-aryl-substituted tripyrranes in the presence of Cu(OTf)2 as the catalyst. The reaction mechanism of the presented method was studied on model reactions by electrospray-ionization time-of-flight (HRESI–TOF) mass spectral analysis in a timely manner. The analytical results indicated that the observed azafulvene-ended di- and tripyrrolic intermediates are responsible for the formation of porphyrinogen and hexaphyrinogen forms.

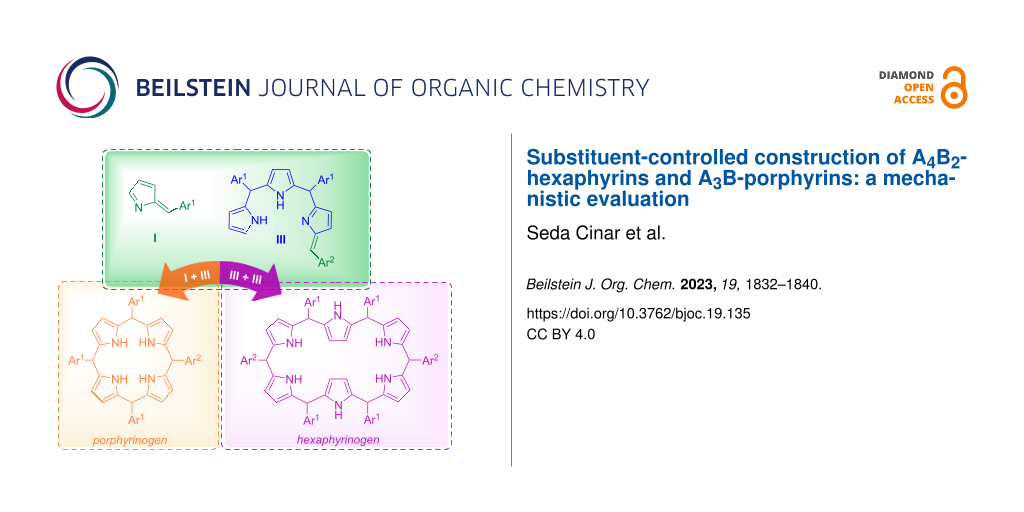
Graphical Abstract
Introduction
Porphyrins and expanded porphyrins have found widespread applications in supramolecular chemistry [1-4]. Expanded porphyrins are utilized as building blocks in the fields of near-infrared (NIR) dyes [5], nonlinear optical (NLO) materials [2], and photosensitizers in photodynamic therapy [1], however, their synthesis is still a challenge for chemists. Hexaphyrins are one of the most investigated structures among expanded porphyrins owing to their structural stability, flexibility, or complexing ability with transition metals [6-12]. meso-Aryl-substituted dipyrromethanes or tripyrranes are the most commonly used starting materials in hexaphyrin syntheses [13-16]. Osuka et al. made significant contributions to the selective synthesis of expanded porphyrins and their chemistry with regard to their aromaticity and coordination properties. They used meso-aryl-substituted dipyrromethanes and aldehydes in the synthesis of A3B3-type hexaphyrins [13] and 5,10-diaryl-substituted tripyrranes in A4B2-hexaphyrin synthesis [6,17-20]. Similarly, the syntheses of AmBn-type hexaphyrins, octaphyrins, or higher expanded porphyrins were handled by improved methods in recent years with the use of tripyrranes or bilanes and aldehydes [7,14,21-25].
In our previous studies, we used N-tosylimines throughout the syntheses of several porphyrinic compounds which emphasized the usability of N-tosylimines with dipyrromethanes, tripyrranes, or bilanes instead of aldehydes in the synthesis of porphyrins and contracted/expanded porphyrins [26-28]. It was shown that the reaction of meso-pentafluorophenyl-substituted N-tosylimine and 5,10-bis(pentafluorophenyl)tripyrromethane formed A6-hexaphyrin as the main product along with the inevitable formation of side products, A4-porphyrin and higher expanded porphyrins [28]. meso-Phenyloligopyrroles having electron-rich substituents at the 2-, 4-, or 6-positions were screened in the literature. To the best of our knowledge, hexaphyrin synthesis from the least substituted aryls appears to be not much studied. In the following study, we focused on the use of less hindered variety of precursors in hexaphyrin and porphyrin synthesis via the Cu(OTf)2-catalyzed reaction of tripyrrane and tosylimine according to the retrosynthetic method given in Scheme 1. Here, we present the substituent-dependent selective construction of A4B2-hexaphyrins and A3B-porphyrins with good yields without the formation of expanded counterparts. Beyond the synthesis, for better understanding of the product formation, mass spectral analyses of model reactions were investigated by time-dependent electrospray-ionization time-of-flight (HRESI–TOF) technique.
![[1860-5397-19-135-i1]](/bjoc/content/inline/1860-5397-19-135-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic method for A4B2-hexaphyrin and A3B-porphyrin synthesis.
Scheme 1: Retrosynthetic method for A4B2-hexaphyrin and A3B-porphyrin synthesis.
Results and Discussion
Reactions of meso-pentafluorophenyl-substituted A2-tripyrrane 1 and N-tosylimines 2 were performed in the presence of Cu(OTf)2 (Table 1). Initially, the unsubstituted phenyl-bearing N-tosylimine 2a was reacted with tripyrrane 1 in CH2Cl2 under previously reported conditions [28], however, the desired hexaphyrin could not been isolated. Under these conditions, only product 3a, which is defined as A3B-type porphyrin was isolated with 15% yield (Table 1, entry 1). Then, we run the reactions of A2-tripyrrane 1 with mesityl-containing tosylimine 2b and 2,6-dichlorobenzylidene-substituted substrate 2c. The desired A4B2-hexaphyrins 4b and 4c were obtained in 17% and 16% yield, respectively (Table 1, entries 2 and 3). Next, to elucidate the role of substituents present in the aromatic part of the N-tosylimines, the monohalogenated N-tosylimines 2d–f and N-tosylimine 2g with a strongly electron-withdrawing CF3 substituent in the 4-position were subjected to the reaction with tripyrrane 1. These para-substituted N-tosylimines provided the A4B2-hexaphyrins 4d–g (Table 1, entries 4–7), with the A4B2-hexaphyrin 4d isolated with 18% yield. The products 4e–g were obtained in 7–10% yield and their formation was corroborated by HRMS spectral analysis (Figures S64–S66 in Supporting Information File 1). In these reactions, the A3B-porphyrins concomitantly formed in yields between 9–17%. When p-methoxy- and p-hydroxy-substituted N-tosylimines 2h and 2i were used in this reaction, substrate 2h gave only the A3B-porphyrin while the imine 2i did not form any product (Table 1, entries 8 and 9). To further evaluate the scope of the reaction, heteroaryl-bearing tosylimines were also tested. The thiophene-substituted tosylimine 2j gave hexaphyrin 4j in 17% yield and porphyrin 3j in 10% yield, whereas the indole-bearing tosylimine gave only A3B-porphyrins but no A4B2-hexaphyrin (Table 1, entries 10 and 11). Signals of trace amounts of A2B2-type porphyrins were detected in the mass spectra of some of the products. 1H NMR analysis of the synthesized hexaphyrins proved that the spectra were in consistence with [26]hexaphyrin aromaticity [29]. Several other metal triflates such as Zn(OTf)2, Gd(OTf)3, and Yb(OTf)3 were also tested as catalysts in the reaction of 4-fluorophenyl-substituted tosylimine 2d and tripyrrane 1 and lower yields of the A4B2-hexaphyrins and A3B-porphyrins were obtained compared to the reaction catalyzed by Cu(OTf)2 (Table S2 in Supporting Information File 1).
Table 1: Synthesis of A3B-porphyrins and A4B2-hexaphyrins.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-135-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | N-Tosylimine 2 | Ar |
Yielda (%)
3a–h,j,k |
Yielda (%)
4b–g,j |
| 1 | a | C6H5 | 15 | – |
| 2 | b | 2,4,6-(CH3)3C6H2 | 17 | 17 |
| 3 | c | 2,6-Cl2-C6H3 | 13 | 16 |
| 4 | d | 4-FC6H4 | 17 | 18 |
| 5 | e | 4-ClC6H4 | 9 | 7b |
| 6 | f | 4-BrC6H4 | 16 | 10b |
| 7 | g | 4-CF3C6H4 | 13 | 10b |
| 8 | h | 4-(OCH3)C6H4 | 22 | – |
| 9 | i | 4-(OH)C6H4 | – | – |
| 10 | j | thiophen-2-yl | 10 | 17 |
| 11 | k | indol-3-yl | 22 | – |
aIsolated yields after flash column chromatography; bidentified by HRMS analysis, NMR spectra could not be recorded due to low solubility.
The synthesis of A3B-porphyrins is effortful and only few studies have been reported involving the use of A3-bilanes [30,31] or dipyrromethane–dicarbinols [32], the modification of A4-porphyrins [33], or the reaction of pyrrole with different aldehydes [34]. In the present work, the applied synthetic method provided the A3B-porphyrins in a single-step reaction from bispentafluorophenyl-substituted tripyrrane 1 and variously substituted N-tosylimines 2 along with the targeted [26]hexaphyrins. Additionally, each reaction was also run with aldehydes to compare the effectiveness of N-tosylimines and aldehydes on this system. In most cases, the yields were lower than those in the reactions with N-tosylimines for both A4B2-hexaphyrins and A3B-porphyrins (Table S1 in Supporting Information File 1).
Until now, we have investigated the effect of substituents present in the aryl substituent of the N-tosylimines on the product formation. At this point, we chose 5,10-bis(4-trifluoromethylphenyl)tripyrromethane (5) as a representative example to investigate the role of the tripyrrane on the reaction. A series of reactions of tripyrrane 5 with tosylimines 2c,d,f,h,l,m were performed. In this case, tripyrrane 5 principally formed A3B-porphyrins (Table 2, entries 1–6) and in some cases A2B2-porphyrins, but disfavored the formation of A4B2-hexaphyrins. As outlined in Table 2, the reactions of N-tosylimines 2d,f,l,m with tripyrrane 5 resulted in the formation of A3B-porphyrins 6b,c,e,f in yields ranging between 12–28%, respectively, where the corresponding A2B2-porphyrins were formed in trace amounts (Table 2, entries 2, 3, 5, and 6). A3B-porphyrin 6a was isolated as the sole product with 13% yield (Table 2, entry 1). In the case of N-tosylimine 2h, the reaction gave A3B-porphyrin 6d and trans-A2B2-porphyrin 7d with 21% and 10% yield, respectively (Table 2, entry 4).
Table 2: Synthesis of AmBn-type porphyrins with electron-deficient tripyrrane 5.
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-135-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | N-Tosylimine | A3B-porphyrin | Yielda (%) | A2B2-porphyrin | Yielda (%) |
| 1 | 2c | 6a | 13 | 7a | – |
| 2 | 2d | 6b | 28 | 7b | traceb |
| 3 | 2f | 6c | 15 | 7c | traceb |
| 4 | 2h | 6d | 21 | 7d | 10 |
| 5 | 2l | 6e | 12 | 7e | traceb |
| 6 | 2m | 6f | 12 | 7f | traceb |
aIsolated yields after flash column chromatography; bidentified by HRMS analysis.
In this work, the role of substituents on tripyrranes and N-tosylimines on product formation has been shown and the synthesis of A3B-porphyrins and a variety of A4B2-hexaphyrins has been achieved. The presence of the bulky electron-withdrawing pentafluorophenyl group in tripyrranes controls the formation of A4B2-hexaphyrins as mentioned by Osuka and Suzuki [13], besides the formation of A3B-porphyrins. On the other hand, electron-withdrawing but less bulky (4-trifluoromethylphenyl) groups on tripyrrane 5 led to predominant formation of the A3B-porphyrin even when it was reacted with mono-, di-, or penta-substituted aryl N-tosylimino substrates (Table 2).
To elucidate the product diversity and to follow the progress of the reaction, a series of mass spectral analysis of the reaction mixture of 4-fluorophenyl-substituted N-tosylimine 2d and tripyrrane 1 has been conducted at 0 °C. Samples were taken from the reaction medium at certain time intervals within 2 hours and examined by ESI LC–MS.
Throughout the high-resolution electrospray-ionization time-of-flight (HRESI–TOF) mass analysis of the reaction mixture at 0 °C, the following peaks were observed: m/z = 246.0366 ([M + H]+ calcd for C11H5F5N, 246.0337), m/z = 857.1468 ([M + Na]+ calcd for C40H25F11N4O2SNa, 857.1415), m/z = 664.1292 ([M + H]+ calcd for C33H17F11N3, 664.1241), m/z = 1134.2119 ([M + Na]+ calcd for C54H37F12N5O4S2Na, 1134.1988), m/z = 963.1694 ([M + Na]+ calcd for C47H28F12N4O2SNa, 963.1634), m/z = 792.1343 ([M + Na]+ calcd for C40H19F12N3Na, 792.1280), corresponding to the intermediates I–VI, respectively (Figure 1, Figures S46 and S47 in Supporting Information File 1). At the very first two minutes of the reaction run at 0 °C no mass signals attributable to reaction intermediates were observed but only signals of the starting materials 1 and 2d both in the negative and positive ion mode. After two minutes, tripyrrane sulfonamide II and azafulvene I mass peaks were observed. Later on, tripyrrolic intermediates III and VI predominated and the mass peak of IV was observed with poor intensity in the spectra (Figure 1 and Figure S47 in Supporting Information File 1).
![[1860-5397-19-135-1]](/bjoc/content/figures/1860-5397-19-135-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Mass spectrum of the reaction mixture of 1 and 2d at 30 min at 0 °C with assigned intermediates (positive ion mode).
Figure 1: Mass spectrum of the reaction mixture of 1 and 2d at 30 min at 0 °C with assigned intermediates (po...
In our previous works, we have shown that the reaction of pyrrole with N-tosylimines leads to pyrrole sulfonamides as the main products [35]. In another work, in the synthesis of dipyrromethane structures, we have proven the formation of azafulvene intermediates by Cu(OTf)2-appended elimination of sulfonamide groups from pyrrolic sulfonamides [36]. Here in this work, during the reaction at 0 °C, intermediates I–VI were detected (Figure 1). The primary intermediates II and IV are formed by the addition of tripyrrane 1 to tosylimine 2d. Further elimination of N-tosyl group(s) from these intermediates gives azafulvene-ended secondary intermediates III, V, and VI. The observed intermediates I–VI having sulfonamide or azafulvene ends are in accordance with our previous findings [26,35,36]. In addition, the observation of azafulvene I could be attributed to the fragmentation of tripyrrane 1, intermediates II or III as proposed in Figure S75 in Supporting Information File 1. These structures (I–VI) could be said to be responsible for the selective formation of porphyrin and hexaphyrin products. When the temperature was increased to rt, porphyrinogen forms of A3B-porphyrin and A4B2-hexapyhrin were predominately observed (Figure S48 in Supporting Information File 1).
According to high-resolution electrospray-ionization time-of-flight (HRESI–TOF) analysis, at the beginning of the reaction, mass peaks of intermediates II and IV arose as a result of tripyrrane 1 addition to N-tosylimine 2d. Further eliminations of sulfonamide groups from II and IV formed the intermediates III, V and VI. A plausible reaction pathway for the formation of A4B2-hexaphyrin and A3B-porphyrin was suggested taking into account the combination of these detected intermediates (Scheme 2). In route I, [3 + 3] reactions of intermediates II or III, at rt form hexaphyrinogen and the subsequent oxidation gives A4B2-hexaphyrin. Similarly, [3 + 1] reactions of intermediates I and II or I and III provide the formation of porphyrinogen and their oxidation gives A3B-porphyrin as indicated in route II. The presence of starting material 1 in the reaction medium can provide the formation of A4B2-hexaphyrin through the reaction with the intermediates IV, V, or VI. These routes are considered as less likely to happen where intermediates II and III dominate the reaction medium according to the mass analysis as stated before. For this reason, in Scheme 2 below, more probable cyclization pathways have been displayed.
![[1860-5397-19-135-i2]](/bjoc/content/inline/1860-5397-19-135-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A suggested reaction pathway for the formation of A4B2-hexaphyrins and A3B-porphyrins.
Scheme 2: A suggested reaction pathway for the formation of A4B2-hexaphyrins and A3B-porphyrins.
A similar LC–MS analysis was made for the reaction of tripyrrane 5 and 4-methoxyphenyl-substituted tosylimine 2h at 0 °C which mainly formed A3B-porphyrins. This time, the primary tosylated intermediates were not detected, instead N-tosyl eliminated azafulvene-ended secondary intermediates VII–XII (Figure 2) were observed, respectively, at m/z = 224.0627 ([M + H]+ calcd for C12H9F3N, 224.0682), m/z = 445.1150 ([M − H]− calcd for C24H15F6N2, 445.1145), m/z = 409.1411 ([M + H]+ calcd for C24H20F3N2O, 409.1522), m/z = 527.1795 ([M + H]+ calcd for C32H26F3N2O2, 527.1941), m/z = 630.1985 ([M − H]− calcd for C36H26F6N3O, 630.1986), and m/z = 750.2364 ([M + H]+ calcd for C44H34F6N3O2, 750.2550) (Figure S49 in Supporting Information File 1). Although the positive ion peaks of tripyrrolic intermediates XI and XII were observed, any hexaphyrin products did form from this set of reactions. Yet, A3B-porphyrins were clearly and selectively formed over A2B2-porphyrins, even the positive ion peaks of dipyrrolic intermediates VIII, IX, and X have been observed (Figure S49 in Supporting Information File 1). A reaction pathway for the predominant formation of A3B-porphyrin considering the reaction of tripyrrane 5 and tosylimine 2h was also proposed and is given in Figure S50 of Supporting Information File 1, in which only the azafulvene-ended intermediates VII–XII were detected.
![[1860-5397-19-135-2]](/bjoc/content/figures/1860-5397-19-135-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Intermediates in the reaction mixture of 5 and 2h at 30 min at 0 °C.
Figure 2: Intermediates in the reaction mixture of 5 and 2h at 30 min at 0 °C.
Conclusion
In conclusion, a set of A4B2-hexaphyrins and A3B-porphyrins were selectively synthesized through the Cu(OTf)2-catalyzed reactions of N-tosylimines and tripyrranes under mild reaction conditions. With these reactions, it has been shown that the C6F5 group led to the formation of hexaphyrin and porphyrins as well as the monosubstituted aryl-bearing N-tosylimines, but a 4-(CF3)C6H4 group led only to the formation of porphyrin compounds.
A mechanistic perspective for the formation of porphyrinic products was acquired via a set of high-resolution mass analyses of selected model reactions. The results indicated that azafulvene-ended tripyrrolic intermediates III, V, and VI or sulfonamide-ended intermediates II and IV along with monopyrrolic fragment I derives the formation of porphyrins and hexaphyrins. This study offers an insight to the design of A4B2-hexaphyrins and A3B-porphyrins by utilizing the substituents on tripyrranes and N-tosylimines.
Experimental
General method: All reagents and solvents were purchased from Sigma-Aldrich, Fisher Scientific, or Acros Organics and were used without further purification. 1H NMR (400 MHz), 13C NMR (100 MHz), and 19F NMR (376 MHz) spectra were recorded on a Bruker 400, Ultra Shield high-performance digital FT-NMR spectrometer. Data for 1H NMR, 13C NMR, and 19F NMR are reported as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, q= quartet, bs = broad singlet, dd = doublet of doublets, td = triplet of doublets, qd = quartet of doublets), coupling constant, number of atoms. UV–vis absorption spectra were recorded on a Mapada Instruments UV3100PC spectrophotometer. Mass spectra were recorded on an Agilent 1200/6210 high-resolution mass time-of-flight (TOF) LC–MS spectrometer. Reactions were followed by thin-layer chromatography (TLC, Kieselgel 60, F254, Merck) with visualization under UV light. Products were purified by silica gel flash column chromatography (0.05–0.63 mm, 230–400 mesh ASTM, E.Merck). N-Tosylimines 2a–m and 5,10-bis(pentafluorophenyl)tripyrromethane (1) were synthesized according to the previously reported literature procedures [35,37].
Synthesis of porphyrin compounds 3a–h,j,k and 4b–g,j
N-Tosylimine 2 (0.090 mmol) and Cu(OTf)2 (0.0090 mmol) were dissolved in CH2Cl2 (0.5 mL) and stirred at room temperature for 30 minutes under N2 atmosphere. To this mixture was added a solution of 5,10-bis(pentafluorophenyl)tripyrromethane (1, 0.090 mmol) in CH2Cl2 (1.5 mL) and the mixture was stirred at rt for 4 h. Afterwards, DDQ (0.180 mmol) was added to this solution and stirred for another 2 h. The resulting solution was eluted through a short silica gel column with EtOAc and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/hexane 1:50) or preparative thin-layer chromatography on silica gel (Silica gel 60, F254, Merck) where applicable to obtain A3B-porphyrins and A4B2-hexaphyrins. Yields of porphyrins 3a–k were between 9–22% and the yields of hexaphyrins 4b–g,j were between 7–18%.
Synthesis of tripyrrane 5
5,10-Bis(4-trifluoromethylphenyl)tripyrromethane (5) was obtained as side product of dipyrromethane synthesis by the condensation of pyrrole and 4-(trifluoromethyl)benzaldehyde. A typical procedure involves 4-(trifluoromethyl)benzaldehyde (28.7 mmol) and pyrrole (143.6 mmol) in 3 mL:197 mL HCl/H2O. The resulting mixture was controlled by TLC and after 4 h, the mixture was extracted with EtOAc (50 mL × 3). The reaction crude was then purified by flash column chromatography (EtOAc/hexane 1:10) to give compound 5 in 20% yield.
Synthesis of porphyrin compounds 6a–f and 7d
N-Tosylimine 2 (0.097 mmol) and Cu(OTf)2 (0.0097 mmol) were dissolved in CH2Cl2 (0.5 mL) and stirred at room temperature for 30 min under N2 atmosphere. To this mixture was then added a solution of 5,10-bis(4-trifluoromethylphenyl)tripyrromethane (5, 0.097 mmol) in CH2Cl2 (1.5 mL) and stirred at rt for 4 h. Afterwards, DDQ (0.195 mmol) was added to this solution and stirred for another 2 h. The resulting solution was eluted through a short silica gel column with EtOAc and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/hexane 1:50) or preparative thin-layer chromatography on silica gel (silica gel 60, F254, Merck) where applicable to obtain A3B-porphyrins 6a–f, and 7d in yields between 10–28%.
Supporting Information
| Supporting Information File 1: Analytical data and copies of spectra. | ||
| Format: PDF | Size: 5.1 MB | Download |
Acknowledgements
The presented work is a part of the Ph.D. thesis entitled “Synthesis of Porphyrins and Expanded Porphyrins from Oligopyrrolic Compounds and Investigatıon of Their Photophysical Properties” written by Seda Cinar, Hacettepe University, Graduate School of Science and Engineering, Beytepe Campus, 06800, Ankara, Turkey.
Funding
SC thanks The Scientific and Technological Research Council of Turkey (TUBITAK) for doctoral scholarship (2211-C Domestic Ph.D. Scholarship Programme for Priority Areas).
References
-
Sessler, J. L.; Miller, R. A. Biochem. Pharmacol. 2000, 59, 733–739. doi:10.1016/s0006-2952(99)00314-7
Return to citation in text: [1] [2] -
Rath, H.; Sankar, J.; PrabhuRaja, V.; Chandrashekar, T. K.; Nag, A.; Goswami, D. J. Am. Chem. Soc. 2005, 127, 11608–11609. doi:10.1021/ja0537575
Return to citation in text: [1] [2] -
Young, S. W.; Qing, F.; Harriman, A.; Sessler, J. L.; Dow, W. C.; Mody, T. D.; Hemmi, G. W.; Hao, Y.; Miller, R. A. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6610–6615. doi:10.1073/pnas.93.13.6610
Return to citation in text: [1] -
Kostas, I. D.; Coutsolelos, A. G.; Charalambidis, G.; Skondra, A. Tetrahedron Lett. 2007, 48, 6688–6691. doi:10.1016/j.tetlet.2007.07.141
Return to citation in text: [1] -
Wu, D.; Descalzo, A. B.; Weik, F.; Emmerling, F.; Shen, Z.; You, X.-Z.; Rurack, K. Angew. Chem., Int. Ed. 2008, 47, 193–197. doi:10.1002/anie.200702854
Return to citation in text: [1] -
Suzuki, M.; Osuka, A. Chem. – Eur. J. 2007, 13, 196–202. doi:10.1002/chem.200601147
Return to citation in text: [1] [2] -
Misra, R.; Kumar, R.; Chandrashekar, T. K.; Joshi, B. S. J. Org. Chem. 2007, 72, 1153–1160. doi:10.1021/jo061861e
Return to citation in text: [1] [2] -
Jasat, A.; Dolphin, D. Chem. Rev. 1997, 97, 2267–2340. doi:10.1021/cr950078b
Return to citation in text: [1] -
Shin, J.-Y.; Kim, K. S.; Yoon, M.-C.; Lim, J. M.; Yoon, Z. S.; Osuka, A.; Kim, D. Chem. Soc. Rev. 2010, 39, 2751–2767. doi:10.1039/b925417j
Return to citation in text: [1] -
Saito, S.; Osuka, A. Angew. Chem., Int. Ed. 2011, 50, 4342–4373. doi:10.1002/anie.201003909
Return to citation in text: [1] -
Ishida, S.-i.; Soya, T.; Osuka, A. Angew. Chem., Int. Ed. 2018, 57, 13640–13643. doi:10.1002/anie.201808513
Return to citation in text: [1] -
Kang, S.; Hayashi, H.; Umeyama, T.; Matano, Y.; Tkachenko, N. V.; Lemmetyinen, H.; Imahori, H. Chem. – Asian J. 2008, 3, 2065–2074. doi:10.1002/asia.200800229
Return to citation in text: [1] -
Suzuki, M.; Osuka, A. Org. Lett. 2003, 5, 3943–3946. doi:10.1021/ol035650x
Return to citation in text: [1] [2] [3] -
Kamimura, Y.; Shimizu, S.; Osuka, A. Chem. – Eur. J. 2007, 13, 1620–1628. doi:10.1002/chem.200601304
Return to citation in text: [1] [2] -
Taniguchi, R.; Shimizu, S.; Suzuki, M.; Shin, J.-Y.; Furuta, H.; Osuka, A. Tetrahedron Lett. 2003, 44, 2505–2507. doi:10.1016/s0040-4039(03)00328-9
Return to citation in text: [1] -
Tanaka, Y.; Shin, J.-Y.; Osuka, A. Eur. J. Org. Chem. 2008, 1341–1349. doi:10.1002/ejoc.200701132
Return to citation in text: [1] -
Suzuki, M.; Osuka, A. Chem. Commun. 2005, 3685–3687. doi:10.1039/b506586k
Return to citation in text: [1] -
Koide, T.; Kashiwazaki, G.; Suzuki, M.; Furukawa, K.; Yoon, M.-C.; Cho, S.; Kim, D.; Osuka, A. Angew. Chem., Int. Ed. 2008, 47, 9661–9665. doi:10.1002/anie.200804570
Return to citation in text: [1] -
Naoda, K.; Mori, H.; Osuka, A. Chem. Lett. 2013, 42, 22–24. doi:10.1246/cl.2013.22
Return to citation in text: [1] -
Naoda, K.; Mori, H.; Oh, J.; Park, K. H.; Kim, D.; Osuka, A. J. Org. Chem. 2015, 80, 11726–11733. doi:10.1021/acs.joc.5b01348
Return to citation in text: [1] -
Mori, H.; Aratani, N.; Osuka, A. Chem. – Asian J. 2012, 7, 1340–1346. doi:10.1002/asia.201100919
Return to citation in text: [1] -
Naoda, K.; Mori, H.; Aratani, N.; Lee, B. S.; Kim, D.; Osuka, A. Angew. Chem., Int. Ed. 2012, 51, 9856–9859. doi:10.1002/anie.201204446
Return to citation in text: [1] -
Sessler, J. L.; Seidel, D.; Bucher, C.; Lynch, V. Tetrahedron 2001, 57, 3743–3752. doi:10.1016/s0040-4020(01)00243-5
Return to citation in text: [1] -
Saito, S.; Osuka, A. Chem. – Eur. J. 2006, 12, 9095–9102. doi:10.1002/chem.200600671
Return to citation in text: [1] -
Tanaka, T.; Osuka, A. Chem. Rev. 2017, 117, 2584–2640. doi:10.1021/acs.chemrev.6b00371
Return to citation in text: [1] -
Temelli, B.; Unaleroglu, C. Tetrahedron 2009, 65, 2043–2050. doi:10.1016/j.tet.2009.01.009
Return to citation in text: [1] [2] -
Aydin, G.; Temelli, B.; Unaleroglu, C. Eur. J. Org. Chem. 2015, 7583–7593. doi:10.1002/ejoc.201501062
Return to citation in text: [1] -
Cinar, S.; Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2014, 55, 544–547. doi:10.1016/j.tetlet.2013.11.101
Return to citation in text: [1] [2] [3] -
Neves, M. G. P. M. S.; Martins, R. M.; Tomé, A. C.; Silvestre, A. J. D.; Silva, A. M. S.; Félix, V.; Cavaleiro, J. A. S.; Drew, M. G. B. Chem. Commun. 1999, 385–386. doi:10.1039/a808952c
Return to citation in text: [1] -
Hatay, I.; Su, B.; Méndez, M. A.; Corminboeuf, C.; Khoury, T.; Gros, C. P.; Bourdillon, M.; Meyer, M.; Barbe, J.-M.; Ersoz, M.; Záliš, S.; Samec, Z.; Girault, H. H. J. Am. Chem. Soc. 2010, 132, 13733–13741. doi:10.1021/ja103460p
Return to citation in text: [1] -
Nowak‐Król, A.; Plamont, R.; Canard, G.; Edzang, J. A.; Gryko, D. T.; Balaban, T. S. Chem. – Eur. J. 2015, 21, 1488–1498. doi:10.1002/chem.201403677
Return to citation in text: [1] -
Rao, P. D.; Dhanalekshmi, S.; Littler, B. J.; Lindsey, J. S. J. Org. Chem. 2000, 65, 7323–7344. doi:10.1021/jo000882k
Return to citation in text: [1] -
Presolski, S. I.; van der Weegen, R.; Wiesfeld, J. J.; Meijer, E. W. Org. Lett. 2014, 16, 1864–1867. doi:10.1021/ol500182z
Return to citation in text: [1] -
Little, R. G. J. Heterocycl. Chem. 1981, 18, 129–133. doi:10.1002/jhet.5570180126
Return to citation in text: [1] -
Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2005, 46, 7941–7943. doi:10.1016/j.tetlet.2005.09.090
Return to citation in text: [1] [2] [3] -
Temelli, B.; Unaleroglu, C. Tetrahedron 2006, 62, 10130–10135. doi:10.1016/j.tet.2006.08.047
Return to citation in text: [1] [2] -
Ka, J.-W.; Lee, C.-H. Tetrahedron Lett. 2000, 41, 4609–4613. doi:10.1016/s0040-4039(00)00672-9
Return to citation in text: [1]
| 35. | Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2005, 46, 7941–7943. doi:10.1016/j.tetlet.2005.09.090 |
| 34. | Little, R. G. J. Heterocycl. Chem. 1981, 18, 129–133. doi:10.1002/jhet.5570180126 |
| 1. | Sessler, J. L.; Miller, R. A. Biochem. Pharmacol. 2000, 59, 733–739. doi:10.1016/s0006-2952(99)00314-7 |
| 2. | Rath, H.; Sankar, J.; PrabhuRaja, V.; Chandrashekar, T. K.; Nag, A.; Goswami, D. J. Am. Chem. Soc. 2005, 127, 11608–11609. doi:10.1021/ja0537575 |
| 3. | Young, S. W.; Qing, F.; Harriman, A.; Sessler, J. L.; Dow, W. C.; Mody, T. D.; Hemmi, G. W.; Hao, Y.; Miller, R. A. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6610–6615. doi:10.1073/pnas.93.13.6610 |
| 4. | Kostas, I. D.; Coutsolelos, A. G.; Charalambidis, G.; Skondra, A. Tetrahedron Lett. 2007, 48, 6688–6691. doi:10.1016/j.tetlet.2007.07.141 |
| 6. | Suzuki, M.; Osuka, A. Chem. – Eur. J. 2007, 13, 196–202. doi:10.1002/chem.200601147 |
| 7. | Misra, R.; Kumar, R.; Chandrashekar, T. K.; Joshi, B. S. J. Org. Chem. 2007, 72, 1153–1160. doi:10.1021/jo061861e |
| 8. | Jasat, A.; Dolphin, D. Chem. Rev. 1997, 97, 2267–2340. doi:10.1021/cr950078b |
| 9. | Shin, J.-Y.; Kim, K. S.; Yoon, M.-C.; Lim, J. M.; Yoon, Z. S.; Osuka, A.; Kim, D. Chem. Soc. Rev. 2010, 39, 2751–2767. doi:10.1039/b925417j |
| 10. | Saito, S.; Osuka, A. Angew. Chem., Int. Ed. 2011, 50, 4342–4373. doi:10.1002/anie.201003909 |
| 11. | Ishida, S.-i.; Soya, T.; Osuka, A. Angew. Chem., Int. Ed. 2018, 57, 13640–13643. doi:10.1002/anie.201808513 |
| 12. | Kang, S.; Hayashi, H.; Umeyama, T.; Matano, Y.; Tkachenko, N. V.; Lemmetyinen, H.; Imahori, H. Chem. – Asian J. 2008, 3, 2065–2074. doi:10.1002/asia.200800229 |
| 32. | Rao, P. D.; Dhanalekshmi, S.; Littler, B. J.; Lindsey, J. S. J. Org. Chem. 2000, 65, 7323–7344. doi:10.1021/jo000882k |
| 1. | Sessler, J. L.; Miller, R. A. Biochem. Pharmacol. 2000, 59, 733–739. doi:10.1016/s0006-2952(99)00314-7 |
| 33. | Presolski, S. I.; van der Weegen, R.; Wiesfeld, J. J.; Meijer, E. W. Org. Lett. 2014, 16, 1864–1867. doi:10.1021/ol500182z |
| 2. | Rath, H.; Sankar, J.; PrabhuRaja, V.; Chandrashekar, T. K.; Nag, A.; Goswami, D. J. Am. Chem. Soc. 2005, 127, 11608–11609. doi:10.1021/ja0537575 |
| 29. | Neves, M. G. P. M. S.; Martins, R. M.; Tomé, A. C.; Silvestre, A. J. D.; Silva, A. M. S.; Félix, V.; Cavaleiro, J. A. S.; Drew, M. G. B. Chem. Commun. 1999, 385–386. doi:10.1039/a808952c |
| 5. | Wu, D.; Descalzo, A. B.; Weik, F.; Emmerling, F.; Shen, Z.; You, X.-Z.; Rurack, K. Angew. Chem., Int. Ed. 2008, 47, 193–197. doi:10.1002/anie.200702854 |
| 30. | Hatay, I.; Su, B.; Méndez, M. A.; Corminboeuf, C.; Khoury, T.; Gros, C. P.; Bourdillon, M.; Meyer, M.; Barbe, J.-M.; Ersoz, M.; Záliš, S.; Samec, Z.; Girault, H. H. J. Am. Chem. Soc. 2010, 132, 13733–13741. doi:10.1021/ja103460p |
| 31. | Nowak‐Król, A.; Plamont, R.; Canard, G.; Edzang, J. A.; Gryko, D. T.; Balaban, T. S. Chem. – Eur. J. 2015, 21, 1488–1498. doi:10.1002/chem.201403677 |
| 7. | Misra, R.; Kumar, R.; Chandrashekar, T. K.; Joshi, B. S. J. Org. Chem. 2007, 72, 1153–1160. doi:10.1021/jo061861e |
| 14. | Kamimura, Y.; Shimizu, S.; Osuka, A. Chem. – Eur. J. 2007, 13, 1620–1628. doi:10.1002/chem.200601304 |
| 21. | Mori, H.; Aratani, N.; Osuka, A. Chem. – Asian J. 2012, 7, 1340–1346. doi:10.1002/asia.201100919 |
| 22. | Naoda, K.; Mori, H.; Aratani, N.; Lee, B. S.; Kim, D.; Osuka, A. Angew. Chem., Int. Ed. 2012, 51, 9856–9859. doi:10.1002/anie.201204446 |
| 23. | Sessler, J. L.; Seidel, D.; Bucher, C.; Lynch, V. Tetrahedron 2001, 57, 3743–3752. doi:10.1016/s0040-4020(01)00243-5 |
| 24. | Saito, S.; Osuka, A. Chem. – Eur. J. 2006, 12, 9095–9102. doi:10.1002/chem.200600671 |
| 25. | Tanaka, T.; Osuka, A. Chem. Rev. 2017, 117, 2584–2640. doi:10.1021/acs.chemrev.6b00371 |
| 28. | Cinar, S.; Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2014, 55, 544–547. doi:10.1016/j.tetlet.2013.11.101 |
| 35. | Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2005, 46, 7941–7943. doi:10.1016/j.tetlet.2005.09.090 |
| 37. | Ka, J.-W.; Lee, C.-H. Tetrahedron Lett. 2000, 41, 4609–4613. doi:10.1016/s0040-4039(00)00672-9 |
| 6. | Suzuki, M.; Osuka, A. Chem. – Eur. J. 2007, 13, 196–202. doi:10.1002/chem.200601147 |
| 17. | Suzuki, M.; Osuka, A. Chem. Commun. 2005, 3685–3687. doi:10.1039/b506586k |
| 18. | Koide, T.; Kashiwazaki, G.; Suzuki, M.; Furukawa, K.; Yoon, M.-C.; Cho, S.; Kim, D.; Osuka, A. Angew. Chem., Int. Ed. 2008, 47, 9661–9665. doi:10.1002/anie.200804570 |
| 19. | Naoda, K.; Mori, H.; Osuka, A. Chem. Lett. 2013, 42, 22–24. doi:10.1246/cl.2013.22 |
| 20. | Naoda, K.; Mori, H.; Oh, J.; Park, K. H.; Kim, D.; Osuka, A. J. Org. Chem. 2015, 80, 11726–11733. doi:10.1021/acs.joc.5b01348 |
| 28. | Cinar, S.; Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2014, 55, 544–547. doi:10.1016/j.tetlet.2013.11.101 |
| 36. | Temelli, B.; Unaleroglu, C. Tetrahedron 2006, 62, 10130–10135. doi:10.1016/j.tet.2006.08.047 |
| 13. | Suzuki, M.; Osuka, A. Org. Lett. 2003, 5, 3943–3946. doi:10.1021/ol035650x |
| 14. | Kamimura, Y.; Shimizu, S.; Osuka, A. Chem. – Eur. J. 2007, 13, 1620–1628. doi:10.1002/chem.200601304 |
| 15. | Taniguchi, R.; Shimizu, S.; Suzuki, M.; Shin, J.-Y.; Furuta, H.; Osuka, A. Tetrahedron Lett. 2003, 44, 2505–2507. doi:10.1016/s0040-4039(03)00328-9 |
| 16. | Tanaka, Y.; Shin, J.-Y.; Osuka, A. Eur. J. Org. Chem. 2008, 1341–1349. doi:10.1002/ejoc.200701132 |
| 26. | Temelli, B.; Unaleroglu, C. Tetrahedron 2009, 65, 2043–2050. doi:10.1016/j.tet.2009.01.009 |
| 27. | Aydin, G.; Temelli, B.; Unaleroglu, C. Eur. J. Org. Chem. 2015, 7583–7593. doi:10.1002/ejoc.201501062 |
| 28. | Cinar, S.; Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2014, 55, 544–547. doi:10.1016/j.tetlet.2013.11.101 |
| 26. | Temelli, B.; Unaleroglu, C. Tetrahedron 2009, 65, 2043–2050. doi:10.1016/j.tet.2009.01.009 |
| 35. | Temelli, B.; Unaleroglu, C. Tetrahedron Lett. 2005, 46, 7941–7943. doi:10.1016/j.tetlet.2005.09.090 |
| 36. | Temelli, B.; Unaleroglu, C. Tetrahedron 2006, 62, 10130–10135. doi:10.1016/j.tet.2006.08.047 |
© 2023 Cinar et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.