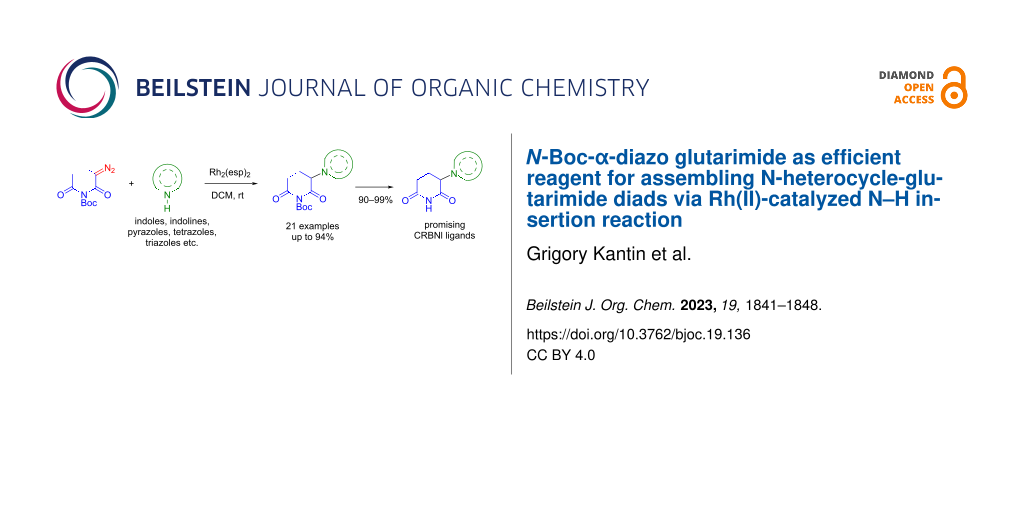
Abstract
A technique has been proposed for incorporating a heterocyclic component into a glutarimide framework employing a Rh2(esp)2-catalyzed N–H insertion with the involvement of N-Boc-α-diazo glutarimide. The new diazo reagent is more stable, soluble and convenient to prepare than the previously suggested one. The approach permits the application of diverse heterocycles, including both aromatic and saturated NH-substrates. This yields structures that are appealing for generating cereblon ubiquitin-ligase ligands and for potential use in crafting PROTAC molecules.

Graphical Abstract
Introduction
Targeted protein degradation (TPD) has transformed the field of drug discovery [1,2]. Utilizing proximity-induced pharmacological strategies [3], this method has fostered the creation of numerous molecular glues and proteolysis-targeting chimeras (PROTACs). By manipulation of the internal ubiquitin-proteasome degradation system, it has been achieved to approach previously considered undruggable targets and conceive remedies for drug-resistant targets [4]. The given TPD's benefit over traditional inhibition; numerous proteins have been set for proteasomal degradation by transmuting accessible small molecule inhibitors into PROTACs. The RAS proteins, once seen as drug-resistant, became susceptible and a series of covalent inhibitors [5-7] were synthesized to bind to KRASG12C. The application of the PROTAC principle has demonstrated the feasibility of endogenous degradation of KRAS [8-10], potentially opening a path for its use in treating KRAS-induced cancers.
A common characteristic of the degraders elaborated in the literature involves the compulsory integration of an E3-ligase ligand motif into the PROTAC configuration [11]. The E3-ligase most frequently utilized in TPD strategies is cereblon (CRBN), the target focus of a collection of immunomodulatory drugs containing the glutarimide moiety such as thalidomide, pomalidomide, lenalidomide [12,13], and avadomide [14] (Figure 1). These ligands, although prevalent recruiters in PROTAC design, present several drawbacks including the degradation of lymphoid transcription factors [15-17] IKZF1, IKZF3, and SALL4 where the latter's degradation could result in a significant teratogenic effect [18]. In addition, these glutarimide derivatives are highly susceptible to hydrolysis and enzymatic cleavage under physiological circumstances, considerably impacting their pharmacological utility [19,20], Moreover, the conventional CRBN recruiters restrict structural modification options necessary to maintain satisfactory affinity for the E3-ligase [21-24]. These limitations underscore the relevance of expanding the chemical space of cereblon ligands.
![[1860-5397-19-136-1]](/bjoc/content/figures/1860-5397-19-136-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Glutarimide-based immunomodulatory drugs (IMiDs) and CRBN ligands.
Figure 1: Glutarimide-based immunomodulatory drugs (IMiDs) and CRBN ligands.
Research teams and pharmaceutical companies worldwide are actively conducting studies to discover new CRBN ligands based on α-hetaryl-substituted glutarimides of general formula 1 (Scheme 1). Consequently, a significant number of patents describing the synthesis of new PROTAC molecules and CRBN ligands are being published (over 400K patents in the last 5 years according to SciFinder). Various nitrogen heterocycles were utilized as a heterocyclic moiety linked to a glutarimide core via a nitrogen atom. In addition to the phthalimide fragment, the most commonly studied ones are pyrazole derivatives (including indazole), benzimidazole, 1,2,3-triazole, indole, carbazole, indoline, quinazoline, and isoquinoline. Nevertheless, many heterocyclic motifs still remain beyond the attention of researchers. For example, glutarimides that incorporate tetrazole and 1,2,4-triazole substituents at the α-position have not yet been studied.
![[1860-5397-19-136-i1]](/bjoc/content/inline/1860-5397-19-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Main literature approaches towards α-hetaryl glutarimides 1 (routes A and B) and new “diazo” methodology based on Rh(II)-catalyzed N–H-insertion reaction.
Scheme 1: Main literature approaches towards α-hetaryl glutarimides 1 (routes A and B) and new “diazo” method...
Typically, two primary strategies are employed to build an N-heterocycle-glutarimide diad. The first involves assembling a heterocyclic fragment utilizing glutamic acid imide (2, Scheme 1, route A), while the second involves alkylating NH-heterocycle with α-bromo (α-oxysulfonyl) glutarimide (3, Scheme 1, route B). Although the first approach has readily available starting reagents, a simple synthesis process, and gives generally high yields, it is restricted in terms of the type of heterocycle that can be assembled (primarily cyclic imides and lactams). The second approach, which asserts its universality, frequently displays inadequate yield of target compounds. As such, the endeavour to discover novel approaches for synthesizing N-heterocycle-glutarimide ensembles of type 1 persists as imperative.
Aliphatic diazocarbonyl reagents are widely acknowledged to be effective in introducing substituents to the nitrogen atoms of NH-heterocycles by means of carbene insertion into the N–H bond upon catalytic or photolytic decomposition of diazo compounds [25]. Furthermore, the reaction of N-heterocycles containing multiple non-equivalent nitrogen atoms with a diazo reagent under neutral conditions may give rise to a separate regioisomer which is different from the product obtained by reaction with an alkyl halide under basic conditions.
Recently, we designed a thalidomide analogue 1a in which the phthalimide moiety was replaced with benzotriazole, using a new synthesis strategy based on the usage of diazo glutarimide 4 (Scheme 1). Compared to thalidomide (Figure 1), the resulting “benzotriazolo thalidomide” has a similar binding mode, but improved properties, as revealed in crystallographic analyses, affinity assays and cell culture [26]. However, the proposed diazo reagent 4 possesses several drawbacks, primarily, insufficient stability during storage and comparatively low solubility in non-polar solvents, as well as complications when isolating it in a pure form.
This study focuses on the development of a "diazo" technique to incorporate a heterocyclic fragment into the glutarimide core, creating potential CRBN ligands and crucial building blocks for assembling PROTACs. In this paper, a more convenient diazo reagent 5 is introduced and its effectiveness in reacting with a broad variety of NH-heterocycles and in providing N-Boc-protected precursors 6 of hetaryl-substituted glutarimides 1 is demonstrated.
Results and Discussion
To address the challenges of working with N-unsubstituted diazo glutarimide 4, a protective and easily removable group was introduced to the molecule. The key diazo reagent 5 was synthesized using modified standard procedures detailed below (Scheme 2). Glutarimide (7), which is commercially available, was (dimethylamino)methylenated at the α-position with the Bredereck's reagent before adding the Boc group to the imide nitrogen atom of the crude enamine. The interim derivative 8 was obtained with a yield of 53% over two steps and readily entered the diazo transfer reaction with 4-nitrophenylsulfonyl azide (4-NsN3). The resulting diazo reagent 5 was produced in a high yield after undergoing straightforward chromatographic purification.
![[1860-5397-19-136-i2]](/bjoc/content/inline/1860-5397-19-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of diazo reagent 5.
Scheme 2: Preparation of diazo reagent 5.
The use of a Boc group at the nitrogen atom of the diazo imide significantly simplified the process of isolating compound 5, as compared to the prior publication [26], and notably increased its stability. Furthermore, the solubility of the new diazo reagent in non-polar solvents, particularly DCM, was significantly improved, leading to a beneficial impact on the course of Rh(II)-catalyzed N–H insertion reactions involving NH-heterocycles. The proposed method enables the synthesis of multigram quantities of diazo compound 5 rapidly. Furthermore, it can be stored up to several weeks in the refrigerator (5 °C) without any observable alterations.
A diverse array of NH-heterocycles with varying characteristics were selected as substrates for the studied insertion reaction, encompassing both aromatic and non-aromatic compounds differing in the number and arrangement of nitrogen atoms (Scheme 3). Notably, several of these heterocycles, including tetrahydroquinoline, 1,2,4-triazole, and tetrazoles, had not been previously utilized in the CRBN ligands design. Catalytic decomposition reactions of diazo compound 5 with NH-heterocycles were conducted in a dry DCM solution using dirhodium espinoate (Rh2(esp)2, 0.06–0.18 mol %). The Rh2(esp)2 catalyst was selected for its excellent versatility and efficiency in various XH insertion reactions [27-29].
![[1860-5397-19-136-i3]](/bjoc/content/inline/1860-5397-19-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of NH insertion reaction of N-Boc-α-diazo glutarimide and various N-heterocycles. aIsolated yield; reaction scale ‒ 0.33 mmol of NH-heterocycle, 0.4 mmol of diazo reagent 5, catalyst ‒ 2.5 mM Rh2(esp)2 in DCM, 100‒300 μL (0.06‒0.18 mol %); bNMR yield; cstructure confirmed by single-crystal X-ray data; dcalculated yield based on incomplete conversion of NH-substrate.
Scheme 3: Scope of NH insertion reaction of N-Boc-α-diazo glutarimide and various N-heterocycles. aIsolated y...
In many cases (e.g., indoles, indazoles, benzotriazoles, tetrazoles, Scheme 3) the reaction progressed expeditiously (as indicated by gas evolution upon adding diazo reagent to the mixture of NH-substrate and catalyst) and was completed in 3–5 h (TLC control). Meanwhile, the disappearance of the diazo reagent was considerably slower in the case of more basic heterocycles (pyrazoles, indolines, tetrahydroquinolines), taking anywhere from 16 to 24 hours, and in some cases (imidazole, 7-azaindazole, ethyl isonipecotate, hexamethylenimine), up to 2–3 days, along with the addition of an extra portion or two of catalyst to complete the reaction. Furthermore, the yields of the NH-insertion products in the latter reactions were moderate or low (see 6h, 6r, 6s, Scheme 3). It should be noted that we have also tested some Cu(II) catalysts (Cu(OTf)2 and Cu(acac)2 in DCE at 80 °C) in reaction with ethyl isonipecotate (see Supporting Information File 1 for details). While in the case of Cu(OTf)2 (5 mol %), the reaction mixture did not contain the desired product 6r, using Cu(acac)2 (10 mol %) resulted in 25% NMR yield of the insertion product. However, this does not show much advantage over Rh2(esp)2 (0.18 mol %), which gave a preparative yield of 18%. It is important to note that despite the higher cost, the rhodium catalyst offers milder reaction conditions.
In the case of some electron-rich substrates, in addition to N–H insertion products 6, C–H insertion products 9 were also observed. Thus, when reacting with indole, the product of carbenoid attack at position 3 (9a) was isolated along with target compound 6a. Introduction of a carbomethoxy group into this position of indole leads to the exceptional formation of the N–H insertion product 6b in high yield. The reaction with methyl pyrrole-2-carboxylate resulted in the isolation of only the C–H insertion product 9c in low yield. Similar reaction progress was observed in the case with imidazole, the product N–H insertion was observed only in trace amounts (according to NMR data of the reaction mixture). The structure of the main reaction product 9i was confirmed by 2D HSQC NMR spectroscopy.
To evaluate the influence of the catalyst on chemoselectivity of the reaction with indole (ratio 6a/9a) we have performed additional testing with Rh2(TFA)4 and Rh2(OAc)4, which differ from the Rh2(esp)2 in both electronic and steric factors (see Supporting Information File 1). When Rh2(OAc)4 (1 mol %) was used, a 1.7:1 ratio of 6a/9a insertion products was observed, which is not much different from the result obtained with Rh2(esp)2. However, the overall yield of insertion products decreased (62% (NMR) vs 69% (isolated)). When Rh2(TFA)4 (1 mol %) was used, a reversal of the ratio of 6a/9a insertion products (1:2) was observed in the test reaction, although the total yield estimated by NMR was only 32%. Thus, in the series of tested Rh(II) catalysts Rh2(esp)2 is the most successful catalyst for obtaining the product of insertion into the NH bond of the heterocycle.
An effort to obtain the N–H insertion product with dibenzoazepine proved fruitless. Instead, the product of insertion into the C–H bond of the activated benzene ring (9z) was isolated in moderate yield. The chemo- and regioselectivity of this reaction can be attributed to several factors, including steric hindrances to the attack of the nitrogen atom by the carbenoid. When reacting with tetrahydroquinoline (with insignificant steric shielding of the N-nucleophilic center), the product of N–H insertion 6x was obtained in good yield. As a side process, а carbenoid's repeated attack at the 6-position of the tetrahydroquinoline ring of compound 6x was observed according to NMR data. Introducing a bromine atom to block the 6-position led to a significant increase in the target product 6y yield.
The decomposition of diazo reagent 5 in the presence of symmetrically substituted pyrazoles produced the relevant products of N–H insertion 6d–f in high yields. The substitution of methyl groups with phenyl groups (6d vs 6e) had no significant impact on the outcome of the reaction. The synthesis of compound 6f was conducted on a gram scale, and enhancing the scale of the reaction did not affect the product yield (93% vs 94%).
Benzimidazole displayed relatively low reactivity, resulting in moderate yield of the desired product 6j. The introduction of a chlorine atom at the 2-position of the substrate (2-chlorobenzimidazole) actively suppressed the targeted reaction. Merely traces of the N–H insertion product were detectable in NMR data, with insignificant conversion of the initial heterocycle.
Examples of N–H insertion reactions with azoles containing non-equivalent nitrogen atoms deserve separate discussion. As the only product of the reaction with indazole, compound 6g, formed as a result of the attack of the carbenoid on the 2-N atom, was obtained. The regioselectivity of the reaction with 7-azaindazole was similar, however, the introduction of an additional basic nitrogen atom resulted in a significant increase in reaction time and a decrease in the yield of the target compound 6h. The structures of the obtained regioisomers 6g and 6h were confirmed by the NOESY spectra, in which cross-peaks between the protons of the glutarimide ring and the singlet of the proton of the pyrazole cycle are observed.
Reactions with benzotriazoles are also highly regioselective, proceeding via attack on the 2-N atom to give products 6m–o. In the instance of unsubstituted benzotriazole, a minor regioisomer 6m' was identified with a calculated yield of 6% using NMR data. Benzotriazoles featuring electron-withdrawing substituents showed incomplete conversion of the NH-heterocycle, and the yields of target compounds 6n and 6o were slightly diminished. The single-crystal X-ray data verified the structure of product 6n. The data obtained on the regioselectivity of reactions with benzotriazoles are in agreement with those previously reported in the literature [30].
Despite the low reactivity (the reaction was carried out for 5 days with triple portion of the catalyst), the yield of the N–H insertion product involving 1,2,4-triazole was unexpectedly high (80%). Compound 6l was obtained as a single regioisomer. The reaction with tetrazoles is also characterized by high regioselectivity. We obtained tetrazol-2-yl glutarimides 6p and 6q in moderate yields. Alternative regioisomers (tetrazol-1-yl glutarimides) were observed only in trace amounts (according to NMR data). It should be noted that in this work the insertion of the diazocarbonyl reagent into the N–H bond of tetrazoles and 1,2,4-triazole was realized for the first time. This transformation can serve as a powerful tool to carry out N-modification of these heterocycles.
Despite all our efforts, it was not possible to obtain N–H insertion products with N-heterocycles containing an α-carbonyl group (Figure 2). We observed formation of complex mixtures in the case of δ-valerolactam, glutarimide and oxindole. This observation can be attributed to the competing direction of the attack of the carbenoid onto the carbonyl oxygen atom resulting in unstable intermediates. In the reaction with 5-phenylpyrazolin-3-one, an exceptionally low conversion of the starting heterocycle was observed.
![[1860-5397-19-136-2]](/bjoc/content/figures/1860-5397-19-136-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Examples of α-carbonyl NH-heterocycles for which N–H insertion products could not be obtained.
Figure 2: Examples of α-carbonyl NH-heterocycles for which N–H insertion products could not be obtained.
In the next step, we have demonstrated the possibility of removing the protective Boc group under mild conditions without acid or base catalysis (Scheme 4). Deprotection occurs in high, near quantitative yields, resulting in glutarimides with a heterocyclic fragment at the α-position 1a–e – structures in demand for the design of CRBN ligands and immunomodulatory drugs.
![[1860-5397-19-136-i4]](/bjoc/content/inline/1860-5397-19-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Examples of N-deprotection of α-modified glutarimides 1.
Scheme 4: Examples of N-deprotection of α-modified glutarimides 1.
In compound 6n, catalytic hydrogenation was used to reduce the nitro group, resulting in the production of a benzotriazole analog of pomalidomide/lenalidomide precursor 10 with a high yield (Scheme 5). In the near future, after removal of the protective group, the biological profile of the compound obtained will be studied (i.e., antimyeloid activity, degradation of transcription factors IKZF1/2/3, antiangiogenic activity, and cytokine secretion), which will be reported in the following works.
![[1860-5397-19-136-i5]](/bjoc/content/inline/1860-5397-19-136-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of NH2-containing derivative 10 via reduction of 6n.
Scheme 5: Preparation of NH2-containing derivative 10 via reduction of 6n.
Conclusion
In summary, a new diazo reagent for the convenient incorporation of heterocyclic substituents at the alpha-position of glutarimide by Rh(II)-catalyzed insertion of carbene into the N–H bond of nitrogen heterocycles has been proposed. The method allows the preparation of modified glutarimides with a wide range of aromatic and aliphatic NH-heterocycles under mild conditions in moderate to high yields. It is shown that electron-rich substrates tend to give C–H insertion products. The N-modification of tetrazoles and 1,2,4-triazoles using a diazocarbonyl reagent is presented for the first time. The protective group is removed without acid catalysis with near quantitative yields. New benzotriazole derivatives containing functional groups capable of participating in the subsequent modification for linker attachment to assemble the PROTAC molecule have been obtained.
Supporting Information
Deposition number 2298240 (for 6n) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service https://www.ccdc.cam.ac.uk/structures.
| Supporting Information File 1: General experimental information, X-ray crystallographic data, synthetic procedures, analytical data and NMR spectra for the reported compounds. | ||
| Format: PDF | Size: 5.7 MB | Download |
References
-
Hanzl, A.; Winter, G. E. Curr. Opin. Chem. Biol. 2020, 56, 35–41. doi:10.1016/j.cbpa.2019.11.012
Return to citation in text: [1] -
Li, K.; Crews, C. M. Chem. Soc. Rev. 2022, 51, 5214–5236. doi:10.1039/d2cs00193d
Return to citation in text: [1] -
Ng, C. S. C.; Banik, S. M. Curr. Opin. Chem. Biol. 2022, 67, 102107. doi:10.1016/j.cbpa.2021.102107
Return to citation in text: [1] -
Samarasinghe, K. T. G.; Crews, C. M. Cell Chem. Biol. 2021, 28, 934–951. doi:10.1016/j.chembiol.2021.04.011
Return to citation in text: [1] -
Chen, C.-y.; Lu, Z.; Scattolin, T.; Chen, C.; Gan, Y.; McLaughlin, M. Org. Lett. 2023, 25, 944–949. doi:10.1021/acs.orglett.2c04266
Return to citation in text: [1] -
Fell, J. B.; Fischer, J. P.; Baer, B. R.; Blake, J. F.; Bouhana, K.; Briere, D. M.; Brown, K. D.; Burgess, L. E.; Burns, A. C.; Burkard, M. R.; Chiang, H.; Chicarelli, M. J.; Cook, A. W.; Gaudino, J. J.; Hallin, J.; Hanson, L.; Hartley, D. P.; Hicken, E. J.; Hingorani, G. P.; Hinklin, R. J.; Mejia, M. J.; Olson, P.; Otten, J. N.; Rhodes, S. P.; Rodriguez, M. E.; Savechenkov, P.; Smith, D. J.; Sudhakar, N.; Sullivan, F. X.; Tang, T. P.; Vigers, G. P.; Wollenberg, L.; Christensen, J. G.; Marx, M. A. J. Med. Chem. 2020, 63, 6679–6693. doi:10.1021/acs.jmedchem.9b02052
Return to citation in text: [1] -
Lorthiois, E.; Gerspacher, M.; Beyer, K. S.; Vaupel, A.; Leblanc, C.; Stringer, R.; Weiss, A.; Wilcken, R.; Guthy, D. A.; Lingel, A.; Bomio-Confaglia, C.; Machauer, R.; Rigollier, P.; Ottl, J.; Arz, D.; Bernet, P.; Desjonqueres, G.; Dussauge, S.; Kazic-Legueux, M.; Lozac’h, M.-A.; Mura, C.; Sorge, M.; Todorov, M.; Warin, N.; Zink, F.; Voshol, H.; Zecri, F. J.; Sedrani, R. C.; Ostermann, N.; Brachmann, S. M.; Cotesta, S. J. Med. Chem. 2022, 65, 16173–16203. doi:10.1021/acs.jmedchem.2c01438
Return to citation in text: [1] -
Bond, M. J.; Chu, L.; Nalawansha, D. A.; Li, K.; Crews, C. M. ACS Cent. Sci. 2020, 6, 1367–1375. doi:10.1021/acscentsci.0c00411
Return to citation in text: [1] -
Yang, F.; Wen, Y.; Wang, C.; Zhou, Y.; Zhou, Y.; Zhang, Z.-M.; Liu, T.; Lu, X. Eur. J. Med. Chem. 2022, 230, 114088. doi:10.1016/j.ejmech.2021.114088
Return to citation in text: [1] -
Zhang, X.; Zhao, T.; Sun, M.; Li, P.; Lai, M.; Xie, L.; Chen, J.; Ding, J.; Xie, H.; Zhou, J.; Zhang, H. Bioorg. Med. Chem. 2023, 78, 117153. doi:10.1016/j.bmc.2023.117153
Return to citation in text: [1] -
Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. Nucleic Acids Res. 2023, 51, D1367–D1372. doi:10.1093/nar/gkac946
Return to citation in text: [1] -
Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Science 2010, 327, 1345–1350. doi:10.1126/science.1177319
Return to citation in text: [1] -
Fischer, E. S.; Böhm, K.; Lydeard, J. R.; Yang, H.; Stadler, M. B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G. M.; Tichkule, R. B.; Schebesta, M.; Forrester, W. C.; Schirle, M.; Hassiepen, U.; Ottl, J.; Hild, M.; Beckwith, R. E. J.; Harper, J. W.; Jenkins, J. L.; Thomä, N. H. Nature 2014, 512, 49–53. doi:10.1038/nature13527
Return to citation in text: [1] -
Heim, C.; Hartmann, M. D. Acta Crystallogr., Sect. D: Struct. Biol. 2022, 78, 290–298. doi:10.1107/s2059798322000092
Return to citation in text: [1] -
Krönke, J.; Udeshi, N. D.; Narla, A.; Grauman, P.; Hurst, S. N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; Ciarlo, C.; Hartman, E.; Munshi, N.; Schenone, M.; Schreiber, S. L.; Carr, S. A.; Ebert, B. L. Science 2014, 343, 301–305. doi:10.1126/science.1244851
Return to citation in text: [1] -
Lu, G.; Middleton, R. E.; Sun, H.; Naniong, M.; Ott, C. J.; Mitsiades, C. S.; Wong, K.-K.; Bradner, J. E.; Kaelin, W. G., Jr. Science 2014, 343, 305–309. doi:10.1126/science.1244917
Return to citation in text: [1] -
Donovan, K. A.; An, J.; Nowak, R. P.; Yuan, J. C.; Fink, E. C.; Berry, B. C.; Ebert, B. L.; Fischer, E. S. eLife 2018, 7, e38430. doi:10.7554/elife.38430
Return to citation in text: [1] -
Matyskiela, M. E.; Couto, S.; Zheng, X.; Lu, G.; Hui, J.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; Lee, C.-W.; Clayton, T.; Fang, W.; Lu, C.-C.; Riley, M.; Abdubek, P.; Blease, K.; Hartke, J.; Kumar, G.; Vessey, R.; Rolfe, M.; Hamann, L. G.; Chamberlain, P. P. Nat. Chem. Biol. 2018, 14, 981–987. doi:10.1038/s41589-018-0129-x
Return to citation in text: [1] -
Bricelj, A.; Dora Ng, Y. L.; Ferber, D.; Kuchta, R.; Müller, S.; Monschke, M.; Wagner, K. G.; Krönke, J.; Sosič, I.; Gütschow, M.; Steinebach, C. ACS Med. Chem. Lett. 2021, 12, 1733–1738. doi:10.1021/acsmedchemlett.1c00368
Return to citation in text: [1] -
Goracci, L.; Desantis, J.; Valeri, A.; Castellani, B.; Eleuteri, M.; Cruciani, G. J. Med. Chem. 2020, 63, 11615–11638. doi:10.1021/acs.jmedchem.0c00793
Return to citation in text: [1] -
Krasavin, M.; Adamchik, M.; Bubyrev, A.; Heim, C.; Maiwald, S.; Zhukovsky, D.; Zhmurov, P.; Bunev, A.; Hartmann, M. D. Eur. J. Med. Chem. 2023, 246, 114990. doi:10.1016/j.ejmech.2022.114990
Return to citation in text: [1] -
Kuchta, R.; Heim, C.; Herrmann, A.; Maiwald, S.; Ng, Y. L. D.; Sosič, I.; Keuler, T.; Krönke, J.; Gütschow, M.; Hartmann, M. D.; Steinebach, C. RSC Chem. Biol. 2023, 4, 229–234. doi:10.1039/d2cb00223j
Return to citation in text: [1] -
Xie, H.; Li, C.; Tang, H.; Tandon, I.; Liao, J.; Roberts, B. L.; Zhao, Y.; Tang, W. J. Med. Chem. 2023, 66, 2904–2917. doi:10.1021/acs.jmedchem.2c01941
Return to citation in text: [1] -
Steinebach, C.; Sosič, I.; Bricelj, A.; Murgai, A.; Bischof, L.; Ng, Y. L. D.; Heim, C.; Maiwald, S.; Proj, M.; Voget, R.; Feller, F.; Košmrlj, J.; Schmidt, A.; Lemnitzer, P.; Hansen, F. K.; Gütschow, M.; Krönke, J.; Hartmann, M. D. ChemRxiv 2023. doi:10.26434/chemrxiv-2023-zshqp
Return to citation in text: [1] -
Solovyev, I. V.; Zhukovsky, D. D.; Dar’in, D. V.; Krasavin, M. Y. Chem. Heterocycl. Compd. 2020, 56, 809–813. doi:10.1007/s10593-020-02736-y
Return to citation in text: [1] -
Krasavin, M.; Bubyrev, A.; Kazantsev, A.; Heim, C.; Maiwald, S.; Zhukovsky, D.; Dar’in, D.; Hartmann, M. D.; Bunev, A. J. Enzyme Inhib. Med. Chem. 2022, 37, 527–530. doi:10.1080/14756366.2021.2024525
Return to citation in text: [1] [2] -
Hunter, A. C.; Chinthapally, K.; Sharma, I. Eur. J. Org. Chem. 2016, 2260–2263. doi:10.1002/ejoc.201600064
Return to citation in text: [1] -
Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565
Return to citation in text: [1] -
Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2020, 3013–3018. doi:10.1002/ejoc.202000067
Return to citation in text: [1] -
Wang, K.; Chen, P.; Ji, D.; Zhang, X.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2018, 57, 12489–12493. doi:10.1002/anie.201807039
Return to citation in text: [1]
| 30. | Wang, K.; Chen, P.; Ji, D.; Zhang, X.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2018, 57, 12489–12493. doi:10.1002/anie.201807039 |
| 1. | Hanzl, A.; Winter, G. E. Curr. Opin. Chem. Biol. 2020, 56, 35–41. doi:10.1016/j.cbpa.2019.11.012 |
| 2. | Li, K.; Crews, C. M. Chem. Soc. Rev. 2022, 51, 5214–5236. doi:10.1039/d2cs00193d |
| 8. | Bond, M. J.; Chu, L.; Nalawansha, D. A.; Li, K.; Crews, C. M. ACS Cent. Sci. 2020, 6, 1367–1375. doi:10.1021/acscentsci.0c00411 |
| 9. | Yang, F.; Wen, Y.; Wang, C.; Zhou, Y.; Zhou, Y.; Zhang, Z.-M.; Liu, T.; Lu, X. Eur. J. Med. Chem. 2022, 230, 114088. doi:10.1016/j.ejmech.2021.114088 |
| 10. | Zhang, X.; Zhao, T.; Sun, M.; Li, P.; Lai, M.; Xie, L.; Chen, J.; Ding, J.; Xie, H.; Zhou, J.; Zhang, H. Bioorg. Med. Chem. 2023, 78, 117153. doi:10.1016/j.bmc.2023.117153 |
| 26. | Krasavin, M.; Bubyrev, A.; Kazantsev, A.; Heim, C.; Maiwald, S.; Zhukovsky, D.; Dar’in, D.; Hartmann, M. D.; Bunev, A. J. Enzyme Inhib. Med. Chem. 2022, 37, 527–530. doi:10.1080/14756366.2021.2024525 |
| 5. | Chen, C.-y.; Lu, Z.; Scattolin, T.; Chen, C.; Gan, Y.; McLaughlin, M. Org. Lett. 2023, 25, 944–949. doi:10.1021/acs.orglett.2c04266 |
| 6. | Fell, J. B.; Fischer, J. P.; Baer, B. R.; Blake, J. F.; Bouhana, K.; Briere, D. M.; Brown, K. D.; Burgess, L. E.; Burns, A. C.; Burkard, M. R.; Chiang, H.; Chicarelli, M. J.; Cook, A. W.; Gaudino, J. J.; Hallin, J.; Hanson, L.; Hartley, D. P.; Hicken, E. J.; Hingorani, G. P.; Hinklin, R. J.; Mejia, M. J.; Olson, P.; Otten, J. N.; Rhodes, S. P.; Rodriguez, M. E.; Savechenkov, P.; Smith, D. J.; Sudhakar, N.; Sullivan, F. X.; Tang, T. P.; Vigers, G. P.; Wollenberg, L.; Christensen, J. G.; Marx, M. A. J. Med. Chem. 2020, 63, 6679–6693. doi:10.1021/acs.jmedchem.9b02052 |
| 7. | Lorthiois, E.; Gerspacher, M.; Beyer, K. S.; Vaupel, A.; Leblanc, C.; Stringer, R.; Weiss, A.; Wilcken, R.; Guthy, D. A.; Lingel, A.; Bomio-Confaglia, C.; Machauer, R.; Rigollier, P.; Ottl, J.; Arz, D.; Bernet, P.; Desjonqueres, G.; Dussauge, S.; Kazic-Legueux, M.; Lozac’h, M.-A.; Mura, C.; Sorge, M.; Todorov, M.; Warin, N.; Zink, F.; Voshol, H.; Zecri, F. J.; Sedrani, R. C.; Ostermann, N.; Brachmann, S. M.; Cotesta, S. J. Med. Chem. 2022, 65, 16173–16203. doi:10.1021/acs.jmedchem.2c01438 |
| 27. | Hunter, A. C.; Chinthapally, K.; Sharma, I. Eur. J. Org. Chem. 2016, 2260–2263. doi:10.1002/ejoc.201600064 |
| 28. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
| 29. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2020, 3013–3018. doi:10.1002/ejoc.202000067 |
| 4. | Samarasinghe, K. T. G.; Crews, C. M. Cell Chem. Biol. 2021, 28, 934–951. doi:10.1016/j.chembiol.2021.04.011 |
| 25. | Solovyev, I. V.; Zhukovsky, D. D.; Dar’in, D. V.; Krasavin, M. Y. Chem. Heterocycl. Compd. 2020, 56, 809–813. doi:10.1007/s10593-020-02736-y |
| 3. | Ng, C. S. C.; Banik, S. M. Curr. Opin. Chem. Biol. 2022, 67, 102107. doi:10.1016/j.cbpa.2021.102107 |
| 26. | Krasavin, M.; Bubyrev, A.; Kazantsev, A.; Heim, C.; Maiwald, S.; Zhukovsky, D.; Dar’in, D.; Hartmann, M. D.; Bunev, A. J. Enzyme Inhib. Med. Chem. 2022, 37, 527–530. doi:10.1080/14756366.2021.2024525 |
| 15. | Krönke, J.; Udeshi, N. D.; Narla, A.; Grauman, P.; Hurst, S. N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; Ciarlo, C.; Hartman, E.; Munshi, N.; Schenone, M.; Schreiber, S. L.; Carr, S. A.; Ebert, B. L. Science 2014, 343, 301–305. doi:10.1126/science.1244851 |
| 16. | Lu, G.; Middleton, R. E.; Sun, H.; Naniong, M.; Ott, C. J.; Mitsiades, C. S.; Wong, K.-K.; Bradner, J. E.; Kaelin, W. G., Jr. Science 2014, 343, 305–309. doi:10.1126/science.1244917 |
| 17. | Donovan, K. A.; An, J.; Nowak, R. P.; Yuan, J. C.; Fink, E. C.; Berry, B. C.; Ebert, B. L.; Fischer, E. S. eLife 2018, 7, e38430. doi:10.7554/elife.38430 |
| 19. | Bricelj, A.; Dora Ng, Y. L.; Ferber, D.; Kuchta, R.; Müller, S.; Monschke, M.; Wagner, K. G.; Krönke, J.; Sosič, I.; Gütschow, M.; Steinebach, C. ACS Med. Chem. Lett. 2021, 12, 1733–1738. doi:10.1021/acsmedchemlett.1c00368 |
| 20. | Goracci, L.; Desantis, J.; Valeri, A.; Castellani, B.; Eleuteri, M.; Cruciani, G. J. Med. Chem. 2020, 63, 11615–11638. doi:10.1021/acs.jmedchem.0c00793 |
| 14. | Heim, C.; Hartmann, M. D. Acta Crystallogr., Sect. D: Struct. Biol. 2022, 78, 290–298. doi:10.1107/s2059798322000092 |
| 21. | Krasavin, M.; Adamchik, M.; Bubyrev, A.; Heim, C.; Maiwald, S.; Zhukovsky, D.; Zhmurov, P.; Bunev, A.; Hartmann, M. D. Eur. J. Med. Chem. 2023, 246, 114990. doi:10.1016/j.ejmech.2022.114990 |
| 22. | Kuchta, R.; Heim, C.; Herrmann, A.; Maiwald, S.; Ng, Y. L. D.; Sosič, I.; Keuler, T.; Krönke, J.; Gütschow, M.; Hartmann, M. D.; Steinebach, C. RSC Chem. Biol. 2023, 4, 229–234. doi:10.1039/d2cb00223j |
| 23. | Xie, H.; Li, C.; Tang, H.; Tandon, I.; Liao, J.; Roberts, B. L.; Zhao, Y.; Tang, W. J. Med. Chem. 2023, 66, 2904–2917. doi:10.1021/acs.jmedchem.2c01941 |
| 24. | Steinebach, C.; Sosič, I.; Bricelj, A.; Murgai, A.; Bischof, L.; Ng, Y. L. D.; Heim, C.; Maiwald, S.; Proj, M.; Voget, R.; Feller, F.; Košmrlj, J.; Schmidt, A.; Lemnitzer, P.; Hansen, F. K.; Gütschow, M.; Krönke, J.; Hartmann, M. D. ChemRxiv 2023. doi:10.26434/chemrxiv-2023-zshqp |
| 12. | Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Science 2010, 327, 1345–1350. doi:10.1126/science.1177319 |
| 13. | Fischer, E. S.; Böhm, K.; Lydeard, J. R.; Yang, H.; Stadler, M. B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G. M.; Tichkule, R. B.; Schebesta, M.; Forrester, W. C.; Schirle, M.; Hassiepen, U.; Ottl, J.; Hild, M.; Beckwith, R. E. J.; Harper, J. W.; Jenkins, J. L.; Thomä, N. H. Nature 2014, 512, 49–53. doi:10.1038/nature13527 |
| 11. | Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. Nucleic Acids Res. 2023, 51, D1367–D1372. doi:10.1093/nar/gkac946 |
| 18. | Matyskiela, M. E.; Couto, S.; Zheng, X.; Lu, G.; Hui, J.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; Lee, C.-W.; Clayton, T.; Fang, W.; Lu, C.-C.; Riley, M.; Abdubek, P.; Blease, K.; Hartke, J.; Kumar, G.; Vessey, R.; Rolfe, M.; Hamann, L. G.; Chamberlain, P. P. Nat. Chem. Biol. 2018, 14, 981–987. doi:10.1038/s41589-018-0129-x |
© 2023 Kantin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.