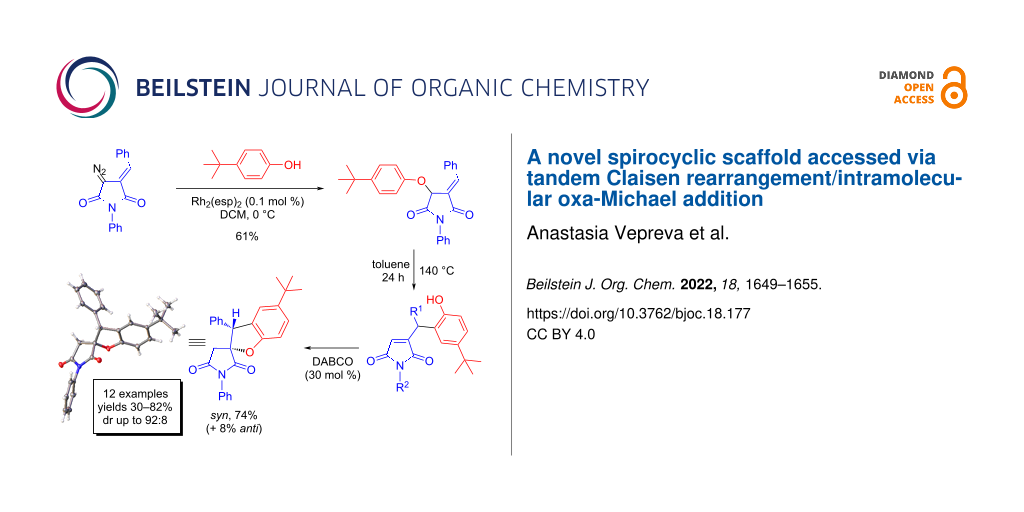
Abstract
A straightforward access to novel spiro[benzofuran-2,3'-pyrrolidine]-2',5'-diones based on the Rh2(esp)2-catalyzed insertion of carbenes derived from diazo arylidene succinimides (DAS) into the O–H bond of phenols is described. The initial adducts underwent a thermally promoted Claisen rearrangement followed by DABCO-catalyzed intramolecular 5-exo-trig oxa-Michael addition.

Graphical Abstract
Introduction
Spirocycles undoubtedly occupy a special place in drug design [1] and, in general, spirocyclic compounds intended for the interrogation of biological targets have been associated with higher success rates [2] in discovering new cases of affinity towards a three-dimensional protein molecule [3]. Spirocycles of all sorts are omnipresent in the natural product realm [4]. Among the approved medicines the following spirocyclic molecules are notable: spironolactone for heart disease and hypertention [5], buspirone for anxiety disorders [6], cevimeline for dry mouth and dry eye syndrome [7], fluspirilene for schizophrenia [8] and ilbesartan for hypertention and diabetic nephropathy [9], to mention a few (Figure 1).
![[1860-5397-18-177-1]](/bjoc/content/figures/1860-5397-18-177-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of approved spirocyclic drugs.
Figure 1: Examples of approved spirocyclic drugs.
Hence the development of novel synthetic methods to construct spirocycles [10-14] constitutes a distinctly worthy undertaking which may as well influence the outlook of the novel medicines discovered and developed in the future.
Recently, we described a novel Rh(II)-catalyzed spirocyclizations involving diazo arylidene succinimide (DAS, such as 1a) with cyclohexanone (as well as other cyclic ketones) which delivered spiro-annulated 2-benzoxepines (such as 2a) along with a minor byproduct 3a identified by 1H NMR as the product of formal insertion of the rhodium(II) carbene species into the O–H bond of cyclohexanone enol form. This minor byproduct, on heating at 50 °C for 12 h, underwent the Claisen rearrangement to give diastereomerically pure maleimide 4a in 4% yield [15]. While for our study at the time the formation of 3 and 4 were viewed as a minor side-reaction, later be started pondering the possibility of giving the observed transformation a stronger impetus from the synthetic point of view. Specifically, we wanted to see if Rh(II)-catalyzed insertion of DAS-derived carbenes could be performed into the O–H bond of phenols and if the resulting phenoxy-substituted succinimides 5 could also undergo a Claisen rearrangement. The products of the latter (6) would again have a reactive phenolic hydroxy group which could potentially be involved in post-condensational transformations such as intramolecular oxa-Michael addition (Scheme 1). Herein, we report our findings obtained in the course of investigating the viability of this strategy.
![[1860-5397-18-177-i1]](/bjoc/content/inline/1860-5397-18-177-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (a) Earlier reported Rh(II)-catalyzed spirocyclization of DAS with the formation of minor enol ether product 3a and its Claisen rearrangement. (b) Synthetic strategy investigated in this work.
Scheme 1: (a) Earlier reported Rh(II)-catalyzed spirocyclization of DAS with the formation of minor enol ethe...
Results and Discussion
The initial attempt to involve DAS 1b in the Rh2(esp)2-catalyzed insertion reaction with 4-(tert-butyl)phenol was successful. The initial adduct 5b was not purified and was heated at 140 °C in toluene to give compound 6b in 47% yield over two steps (Scheme 2). No further optimization of the reaction conditions was undertaken except for the isolation for the initial O–H insertion product which facilitated subsequent steps (the Claisen rearrangement and the final cyclization, vide infra).
![[1860-5397-18-177-i2]](/bjoc/content/inline/1860-5397-18-177-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Initial attempt at Rh(II)-catalyzed O–H insertion/Claisen rearrangement.
Scheme 2: Initial attempt at Rh(II)-catalyzed O–H insertion/Claisen rearrangement.
With product 6a at hand we proceeded studying its base-promoted Michael-type cyclizations. As it follows from the data collated in Table 1, to our delight, cyclizations of the phenoxide anion generated on the action of base (except for 2,6-lutidine) proceeded as 5-exo-trig (rather than 6-endo-trig) process and yielded spirocyclic compound 7a as a mixture of syn and anti diastereomers. DABCO in toluene at room temperature over 1 h (Table 1, entry 5) gave a superior result both in terms of the isolated yield and diastereoselectivity. Increasing the polarity of the solvent appeared to be detrimental to the reaction outcome.
Table 1: Investigation of base-catalyzed transformation of compound 6a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-177-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Solvent | Base (30 mol %) | T (°C) | Total yield (%) | syn/anti | Conversion within 1 h (%) |
| 1 | toluene | DABCO | 0 | 34 | 79:21 | 70 |
| 2 | toluene | DBU | 25 | 63 | 62:38 | 100 |
| 3 | toluene | Cs2CO3 | 25 | 66 | 73:27 | 100 |
| 4 | toluene | 2,6-lutidine | 25 | – | – | – |
| 5 | toluene | DABCO | 25 | 71 | 81:19 | 100 |
| 6 | acetonitrile | DABCO | 25 | 66 | 77:23 | 100 |
| 7 | methanol | DABCO | 25 | 48 | 46:54 | 100 |
| 8 | MeOH/H2O (1:1) | DABCO | 25 | – | – | – |
With the conditions identified for the Rh2(esp)2-catalyzed insertion into the phenolic O–H bond, the Claisen rearrangement and the base-catalyzed cyclization to the spirocyclic product, we experimented with attempts to perform all three steps in a one-pot format. This gave inferior results in terms of product yield and purity. The best result was obtained by performing the Rh(II)-catalyzed insertion first, purifying the respective product 5 and then engaging it in the sequential Claisen rearrangement cyclization reactions performed in one-pot format.
To expand the scope of the newly discovered transformations, several products of the Rh(II)-catalyzed O–H insertions 5 were synthesized as detailed in Scheme 3.
![[1860-5397-18-177-i3]](/bjoc/content/inline/1860-5397-18-177-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Rh2(esp)2-catalyzed O–H insertion reactions between various DAS 1 and phenols.
Scheme 3: Rh2(esp)2-catalyzed O–H insertion reactions between various DAS 1 and phenols.
The O–H insertion reaction worked well for electron-neutral or electron-rich phenols. The presence of chlorine substituents (such as 4-Cl or 2,4-diCl) in the phenol component drastically diminished the yield and led to the formation of the earlier reported DAS dimer [16]. Similarly, electron-donating groups (such as 4-methoxy) in the arylidene portion of 1 complicated the course of the reaction.
Having amassed a sizable arsenal of O–H insertion products 5a–n, we proceeded to study their behavior in the two-step, one-pot sequence of the Claisen rearrangement/intramolecular Michael-type spirocyclization as detailed in Scheme 4. In two cases (7a and 7c), the major (syn) and minor (anti) diastereomers were separated chromatographically and characterized. In one case (7a), the structure of the major (syn) diastereomer was unequivocally confirmed by single-crystal X-ray crystallography. In all other cases, only the pure syn diastereomer was isolated and characterized. The yields of spirocyclic products were generally modest to good over two steps. An electron-accepting group in the benzylidene portion (5j) or an N-benzyl substitution in the starting material (5g) lowered the reactivity and the Claisen rearrangement step was performed at a higher (150 °C) temperature.
![[1860-5397-18-177-i4]](/bjoc/content/inline/1860-5397-18-177-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Two-step, one-pot sequence of the Claisen rearrangement/intramolecular Michael-type spirocyclization of substrates 5a–l. The Claisen rearrangement product was not isolated. Scale from 0.3 to 0.9 mmol; pure major syn diastereomer was isolated and characterized in all cases; ain these examples, pure minor anti diastereomer was isolated and characterized; bthe structure was confirmed by crystallography. csyn/anti ratio is shown in parentheses; dthe reaction was performed at 150 °C.
Scheme 4: Two-step, one-pot sequence of the Claisen rearrangement/intramolecular Michael-type spirocyclizatio...
Notable was our inability to involve o-methoxy- (5n) and (p-methoxy)phenoxy (5m) substrates in the two-step synthesis of the respective spirocycles 7. In both cases, 1H NMR analysis of the reaction mixture indicated the formation of complex product mixtures at the Claisen rearrangement step.
3,5-Dimethylphenoxy-substituted substrate 5h gave the best diastereomeric ratio in the series. The preference for the formation of the syn diastereomer in each case (and for 5a→7a in particular) can, in principle, be rationalized by the greater steric repulsion in the conformer of the respective oxyanion leading to the anti diastereomer compared to that from which the major, syn diastereomer is formed (Scheme 5).
![[1860-5397-18-177-i5]](/bjoc/content/inline/1860-5397-18-177-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Tentative rationalization of the diastereoselectivity observed in all 5→7 transformations (shown for 5a→7a).
Scheme 5: Tentative rationalization of the diastereoselectivity observed in all 5→7 transformations (shown fo...
Notably, a compound analogous to 5a was synthesized with thiophenol in 78% yield. However, it turned out to be completely unreactive towards the Claisen rearrangement step, even at 150 °C in 1,2-dichlorobenzene. Raising the temperature to 200 °C led to starting material deterioration and was not productive either.
Spirocyclic products 7a–l were tested for their ability to influence the survival of MDA-MB-231 (breast adenocarcinoma) and NCI-H460 (lung cancer) cell lines and proved completely non-cytotoxic. This validates these new compounds as suitable molecular probes for interrogating various biological targets via screening in cell-based assays.
Conclusion
We have developed a straightforward access to novel spiro[benzofuran-2,3'-pyrrolidine]-2',5'-diones based on Rh2(esp)2-catalyzed insertion of carbenes derived from α-diazosuccinimides (DAS) into the O–H bond of phenols. The initial adducts underwent a thermally promoted Claisen rearrangement followed by a DABCO-catalyzed intramolecular 5-exo-dig Michael addition. The resulting spirocyclic compounds are formed with a clear preference to the syn diastereomer over anti which can be rationalized by conformational analysis of the Claisen rearrangement precursors to their formation.
Supporting Information
Deposition number 2166113 (for syn-7a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service http://www.ccdc.cam.ac.uk/structures.
| Supporting Information File 1: General experimental information, X-ray crystallographic data, synthetic procedures, analytical data and NMR spectra for the reported compounds. | ||
| Format: PDF | Size: 3.5 MB | Download |
References
-
Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. J. Med. Chem. 2021, 64, 150–183. doi:10.1021/acs.jmedchem.0c01473
Return to citation in text: [1] -
Zheng, Y.; Tice, C. M.; Singh, S. B. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. doi:10.1016/j.bmcl.2014.06.081
Return to citation in text: [1] -
Zheng, Y.-J.; Tice, C. M. Expert Opin. Drug Discovery 2016, 11, 831–834. doi:10.1080/17460441.2016.1195367
Return to citation in text: [1] -
Chupakhin, E.; Babich, O.; Prosekov, A.; Asyakina, L.; Krasavin, M. Molecules 2019, 24, 4165. doi:10.3390/molecules24224165
Return to citation in text: [1] -
Garthwaite, S. M.; McMahon, E. G. Mol. Cell. Endocrinol. 2004, 217, 27–31. doi:10.1016/j.mce.2003.10.005
Return to citation in text: [1] -
Loane, C.; Politis, M. Brain Res. 2012, 1461, 111–118. doi:10.1016/j.brainres.2012.04.032
Return to citation in text: [1] -
Ono, M.; Takamura, E.; Shinozaki, K.; Tsumura, T.; Hamano, T.; Yagi, Y.; Tsubota, K. Am. J. Ophthalmol. 2004, 138, 6–17. doi:10.1016/j.ajo.2004.02.010
Return to citation in text: [1] -
van Epen, J. H. Psychiatr., Neurol., Neurochir. 1970, 73, 277–284.
Return to citation in text: [1] -
Fischer, J.; Robin, G. C. Analogue-based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2006; p 470. doi:10.1002/3527608001
Return to citation in text: [1] -
Ammon, E.; Villinger, A.; Ehlers, P.; Langer, P. Synlett 2022, 33, 1633–1636. doi:10.1055/s-0042-1751363
Return to citation in text: [1] -
Iaroshenko, V. O.; Dudkin, S.; Sosnovskikh, V. Ya.; Villinger, A.; Langer, P. Synthesis 2013, 45, 971–977. doi:10.1055/s-0032-1318273
Return to citation in text: [1] -
Attanasi, O. A.; Favi, G.; Giorgi, G.; Mantellini, F.; Karapetyan, V.; Langer, P. Tetrahedron 2009, 65, 5456–5461. doi:10.1016/j.tet.2009.04.018
Return to citation in text: [1] -
Rasool, N.; Rashid, M. A.; Adeel, M.; Görls, H.; Langer, P. Tetrahedron Lett. 2008, 49, 2254–2257. doi:10.1016/j.tetlet.2008.02.036
Return to citation in text: [1] -
Rasool, N.; Rashid, M. A.; Reinke, H.; Fischer, C.; Langer, P. Tetrahedron 2008, 64, 3246–3252. doi:10.1016/j.tet.2008.01.066
Return to citation in text: [1] -
Vepreva, A.; Kantin, G.; Krasavin, M.; Dar’in, D. Synthesis 2022, 54, 5128–5138. doi:10.1055/s-0037-1610790
Return to citation in text: [1] -
Vepreva, A.; Bunev, A. S.; Kudinov, A. Yu.; Kantin, G.; Krasavin, M.; Dar’in, D. Beilstein J. Org. Chem. 2022, 18, 533–538. doi:10.3762/bjoc.18.55
Return to citation in text: [1]
| 1. | Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. J. Med. Chem. 2021, 64, 150–183. doi:10.1021/acs.jmedchem.0c01473 |
| 5. | Garthwaite, S. M.; McMahon, E. G. Mol. Cell. Endocrinol. 2004, 217, 27–31. doi:10.1016/j.mce.2003.10.005 |
| 4. | Chupakhin, E.; Babich, O.; Prosekov, A.; Asyakina, L.; Krasavin, M. Molecules 2019, 24, 4165. doi:10.3390/molecules24224165 |
| 3. | Zheng, Y.-J.; Tice, C. M. Expert Opin. Drug Discovery 2016, 11, 831–834. doi:10.1080/17460441.2016.1195367 |
| 2. | Zheng, Y.; Tice, C. M.; Singh, S. B. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. doi:10.1016/j.bmcl.2014.06.081 |
| 9. | Fischer, J.; Robin, G. C. Analogue-based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2006; p 470. doi:10.1002/3527608001 |
| 15. | Vepreva, A.; Kantin, G.; Krasavin, M.; Dar’in, D. Synthesis 2022, 54, 5128–5138. doi:10.1055/s-0037-1610790 |
| 16. | Vepreva, A.; Bunev, A. S.; Kudinov, A. Yu.; Kantin, G.; Krasavin, M.; Dar’in, D. Beilstein J. Org. Chem. 2022, 18, 533–538. doi:10.3762/bjoc.18.55 |
| 7. | Ono, M.; Takamura, E.; Shinozaki, K.; Tsumura, T.; Hamano, T.; Yagi, Y.; Tsubota, K. Am. J. Ophthalmol. 2004, 138, 6–17. doi:10.1016/j.ajo.2004.02.010 |
| 6. | Loane, C.; Politis, M. Brain Res. 2012, 1461, 111–118. doi:10.1016/j.brainres.2012.04.032 |
| 10. | Ammon, E.; Villinger, A.; Ehlers, P.; Langer, P. Synlett 2022, 33, 1633–1636. doi:10.1055/s-0042-1751363 |
| 11. | Iaroshenko, V. O.; Dudkin, S.; Sosnovskikh, V. Ya.; Villinger, A.; Langer, P. Synthesis 2013, 45, 971–977. doi:10.1055/s-0032-1318273 |
| 12. | Attanasi, O. A.; Favi, G.; Giorgi, G.; Mantellini, F.; Karapetyan, V.; Langer, P. Tetrahedron 2009, 65, 5456–5461. doi:10.1016/j.tet.2009.04.018 |
| 13. | Rasool, N.; Rashid, M. A.; Adeel, M.; Görls, H.; Langer, P. Tetrahedron Lett. 2008, 49, 2254–2257. doi:10.1016/j.tetlet.2008.02.036 |
| 14. | Rasool, N.; Rashid, M. A.; Reinke, H.; Fischer, C.; Langer, P. Tetrahedron 2008, 64, 3246–3252. doi:10.1016/j.tet.2008.01.066 |
© 2022 Vepreva et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.