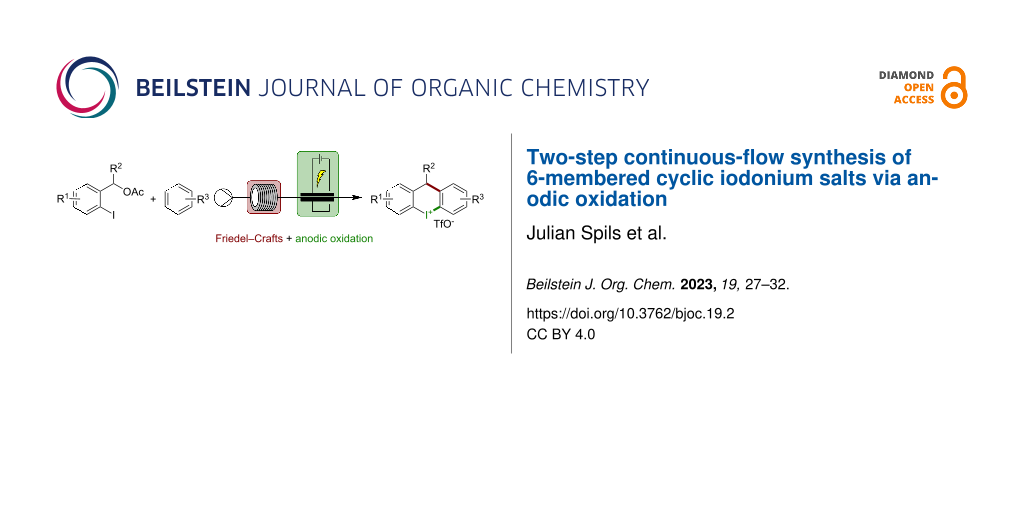
Abstract
We describe a multi-step continuous-flow procedure for the generation of six-membered diaryliodonium salts. The accompanying scalability and atom economy are significant improvements to existing batch methods. Benzyl acetates are submitted to this two-step procedure as highly available and cheap starting materials. An acid-catalyzed Friedel–Crafts alkylation followed by an anodic oxidative cyclization yielded a defined set of cyclic iodonium salts in a highly substrate-dependent yield.

Graphical Abstract
Introduction
Hypervalent iodine compounds (HVI) are well-established reagents for synthetic chemists. They are portrayed as an alternative to otherwise hazardous transition metals. This is due to their great reactivity in electrophilic group transfers [1-4], photo- or organocatalysis [5-15], and their utility as building blocks for the synthesis of natural products [16-21]. One subclass of HVIs is diaryliodonium salts (DIS), which have been used as versatile electrophilic arylation reagents in metal-catalyzed and metal-free reactions [22,23]. The corresponding cyclic diaryliodonium salts (CDIS) have also been investigated as useful building blocks for the synthesis of larger diaryl-based molecules, (hetero)aromatic tricyclic systems, or new aryl-moieties [24,25]. Most methods that generate DIS utilize iodoarenes as starting materials [26,27]. In these one-pot procedures, iodoarenes react with nucleophilic arenes under oxidative acidic conditions.The synthesis of CDIS is more challenging since the arene moiety must be covalently connected to the iodoarene prior to cyclization. Recently, we published two new methods that describe the generation of carbon- and heteroatom-bridged CDIS [28,29]. Herein, we improved the formation of iodoarenes through a Brønsted acid-mediated Friedel–Crafts reaction followed by an oxidative cyclization to form the desired CDIS 1 (Scheme 1A). This one-pot approach is based on ortho-iodinated benzyl alcohols as starting materials. It allows access to a variety of otherwise tedious to synthesize CDIS robustly in short reaction times. A significant drawback still is the use of stoichiometric amounts of chemical oxidants, which decreases the atom economy and necessitates additional workup procedures. A possible solution is the anodic oxidation of iodoarenes as electrochemistry is a highly economical tool that avoids chemical oxidants for synthesizing hypervalent iodine reagents [30]. Iodoarenes are suitable and well-established mediators in either in- or ex-cell electrochemical processes [31-36].
![[1860-5397-19-2-i1]](/bjoc/content/inline/1860-5397-19-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of acyclic (DIS) and cyclic (CDIS) diaryliodonium salts.
Scheme 1: Synthesis of acyclic (DIS) and cyclic (CDIS) diaryliodonium salts.
Nonetheless, HVIs, DIS and CDIS have been generated by anodic oxidation [37-40]. Due to the apparent advantages of electrochemical processes, their implementation in flow is simple and straightforward since further dilution or additives are unnecessary [41,42]. One early example of this combination in the field of HVI chemistry is the anodic oxidation of iodoarenes to form DIS by Wirth et al. (Scheme 1B) [39]. Herein, established conditions for synthesizing DIS were transferred into flow chemistry utilizing a model flow reactor with two platinum electrodes. Other recent examples include the generation of five-membered CDIS utilizing fluorinated alcohols as a solvent [37,38]. Therefore, we attempt to transfer our already established one-pot procedure towards CDIS 1 into a multi-step electrochemical flow process, improving reaction times, atom economy, and scalability (Scheme 1C).
Results and Discussion
We initially investigated the second step of the proposed multi-step procedure since we assumed it to be the more challenging. We initially intended to oxidize and cyclize the intermediate iodoarene 2 electrochemically under batch conditions (Table 1). Through preliminary observations (see Supporting Information File 1, Table S1), triflic acid was established to be suitable for cyclization and as a counterion. 2,2,2-Trifluoroethanol (TFE) as a solvent already resulted in a yield of 45% of compound 1a (Table 1, entry 1). It was possible to reduce its amount to 20 vol % by replacement with MeCN or CH2Cl2 without any significant drop in the yields (40–46%, Table 1, entries 2 and 3). By exchanging TFE for the more stabilizing 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) it was possible to increase the yield even further to 78% (Table 1, entry 4) [43,44]. Finally, we decreased the amount of TfOH to 2 equiv still yielding product 1a in 76% yield (Table 1, entry 5).
Table 1: Optimization of the oxidation and cyclization in batch.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-2-i2.svg?max-width=637&scale=1.0)
|
||||
| Entrya | TfOH [equiv] | Solvent | Charge [F] | 1a Yield [%] |
| 1 | 5 | TFE | 2.1 | 45 |
| 2 | 5 | MeCN/TFE (4:1) | 2.0 | 40 |
| 3 | 5 | CH2Cl2/TFE (4:1) | 2.2 | 46 |
| 4 | 5 | CH2Cl2/HFIP (4:1) | 2.0 | 78 |
| 5 | 2 | CH2Cl2/HFIP (4:1) | 2.0 | 76 |
aElectrolysis was carried out with an ElectraSyn 2.0 in an undivided cell. Vials were equipped with a glassy carbon (GC) anode and a Pt cathode. Immersed area: 2.4 cm2. General reaction conditions: 2 (0.200 mmol), TfOH, TBABF4 (0.005 M), solvent (40.0 mM, 5 mL), 10 mA, CCE.
After establishing the optimized reaction conditions for batch, we wanted to find conditions for a flow procedure. It was observed that plastic parts made out of PEEK did not tolerate the acidic conditions in HFIP, and the solvent was changed back to TFE. This worked surprisingly well with only two equivalents of TfOH forming product 1a in 74% yield (Table 2, entry 1). We found that these reaction conditions cannot be applied in the proposed multi-step procedure since the added benzene clogs the flow reactor through precipitation of a black solid.
Table 2: Optimization of the oxidation and cyclization in flow.
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-2-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | TfOH [equiv] | Additives | Solvent | Current [mA] | 1a Yield [%] |
| 1 | 2 | TBABF4 | CH2Cl2/TFE | 32 (2.0 F) | 74 |
| 2 | 2 |
TBABF4
PhH (9 equiv) |
MeNO2 | 32 (2.0 F) | 48 |
| 3 | 2 |
TBABF4
PhH (9 equiv) |
MeNO2/TFE | 32 (2.0 F) | 63 |
| 4 | 2 | PhH (9 equiv) | MeNO2/TFE | 32 (2.0 F) | 62 |
| 5 | 2 | PhH (9 equiv) | MeNO2/TFE | 48 (3.0 F) | 62 |
aReaction was performed in a Vapourtec Ion electrochemical flow reactor with a glassy carbon (GC) anode and a Pt cathode. General reaction conditions: 1 (0.1 M), TBABF4 (0.005 M). Flowrate: 0.1 mL·min−1. Yield is based on collecting for 20 min (0.200 mmol) after two reactor volumes had passed at the respective conditions.
We could overcome this issue by employing MeNO2 as a solvent, which leads to a 48% yield of 1a (Table 2, entry 2). This yield could be increased further by a combination with TFE to 63% (Table 2, entry 3). We could also omit the additional electrolyte TBABF4 with no product loss (Table 2, entry 4), while the yield could not be improved further by increasing the charge (Table 2, entry 5).
For a final combination of the two reaction steps, benzyl acetate 3a was chosen as a model substrate due to its high solubility and good reactivity as was previously demonstrated by us (Table 3) [28]. We determined an optimal reaction time of 20 min for the Friedel–Crafts reaction via GC analysis. Both steps in series resulted in successful product formation with an albeit slightly diminished yield of 49% (Table 3, entry 3).
Table 3: Optimization of the two-step flow process in series.
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-2-i4.svg?max-width=637&scale=1.0)
|
|||
| Entrya | Electrolyte | Current [mA] | 1a Yield [%] |
| 1 | – | 32 (2.0 F) | 49 |
| 2 | TBABF4 | 32 (2.0 F) | 54 |
| 3 | TBABF4 | 48 (3.0 F) | 54 |
aReaction was performed in a Vapourtec Ion electrochemical flow reactor with a glassy carbon (GC) anode and a Pt cathode. Yield is based on collecting for 20 min (0.200 mmol) after two reactor volumes had passed at the respective conditions.
We increased the yield to 54% by introducing an electrolyte (Table 3, entry 2). A further current increase had no beneficial effect (Table 3, entry 3). We then investigated longer reaction times (4 × 50 min, 4 × 0.500 mmol), but without additional electrolyte the overall yield of 38% was significantly lower. It was possible to compensate for this to some degree by using TBABF4 as shown in Table 3, entry 2. This adjustment improved the overall yield to 43%.
Finally, we investigated a variety of substituted benzyl acetates 3 (Figure 1). Substitutions para to the iodine led with F- and Cl-derivatives 1b and 1c to strongly diminished yields of 15% and 26%, respectively, due to expected lower yields in the Friedel–Crafts step. It was impossible to improve this significantly by increasing the time or temperature. In case of the more electron-rich derivative 1d, we increased the yield to 54%. Other electron-withdrawing substituents show the same trend as in the Br-substituted 1e yielding only 16%. Methylated substrate 1f was isolated in 34% yield indicating that steric effects play a role during the oxidation step. We further wanted to investigate different arenes, but due to the lack of selectivity of the primary benzyl acetates in the Friedel–Crafts step, we were limited to para-substituted arenes, which unfortunately all resulted in the formation of insoluble intermediary iodoarenes. Derivatizing the benzylic position was done by employing secondary benzyl alcohols. These are well soluble and lead to an about 10-times shortened Friedel–Crafts step at 0 °C for the conversion of 3g. Longer times only resulted in the decomposition of the intermediary iodoarene. Here it was possible to perform the procedure with either benzene or toluene, leading to a moderate yield of 38% and 37%. We could not use derivatives with any other substituents with secondary benzyl acetates since those led only to inseparable product mixtures.
![[1860-5397-19-2-1]](/bjoc/content/figures/1860-5397-19-2-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Substrate scope using the optimized conditions of Table 3. Yield is based on collecting for 3 h 20 min (2.00 mmol) after two reactor volumes had passed at the respective conditions. aSolutions of benzyl alocohol and TfOH were precooled in flow before mixing (see Supporting Information File 1).
Figure 1: Substrate scope using the optimized conditions of Table 3. Yield is based on collecting for 3 h 20 min (2....
Conclusion
In summary, we have developed the first multi-step continuous-flow procedure for the generation of cyclic six-membered diaryliodonium salts. Starting from easily accessible benzyl acetates we were able to combine a Friedel–Crafts alkylation with a subsequent anodic oxidative cyclization in flow. The method is currently limited by the narrow starting materials being used due to the rather harsh conditions of those reactions. By addressing this issue, other substrates and higher yields could be realized in future.
Supporting Information
| Supporting Information File 1: Experimental, analytical data and copies of NMR spectra. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h
Return to citation in text: [1] -
Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Caspers, L. D.; Nachtsheim, B. J. Chem. – Asian J. 2018, 13, 1231–1247. doi:10.1002/asia.201800102
Return to citation in text: [1] -
Baralle, A.; Fensterbank, L.; Goddard, J.-P.; Ollivier, C. Chem. – Eur. J. 2013, 19, 10809–10813. doi:10.1002/chem.201301449
Return to citation in text: [1] -
Flores, A.; Cots, E.; Bergès, J.; Muñiz, K. Adv. Synth. Catal. 2019, 361, 2–25. doi:10.1002/adsc.201800521
Return to citation in text: [1] -
Le Vaillant, F.; Garreau, M.; Nicolai, S.; Gryn'ova, G.; Corminboeuf, C.; Waser, J. Chem. Sci. 2018, 9, 5883–5889. doi:10.1039/c8sc01818a
Return to citation in text: [1] -
Wang, D.; Mao, J.; Zhu, C. Chem. Sci. 2018, 9, 5805–5809. doi:10.1039/c8sc01763h
Return to citation in text: [1] -
Boelke, A.; Nachtsheim, B. J. Adv. Synth. Catal. 2019, 362, 184–191. doi:10.1002/adsc.201901356
Return to citation in text: [1] -
Cots, E.; Flores, A.; Romero, R. M.; Muñiz, K. ChemSusChem 2019, 12, 3028–3031. doi:10.1002/cssc.201900360
Return to citation in text: [1] -
Declas, N.; Le Vaillant, F.; Waser, J. Org. Lett. 2019, 21, 524–528. doi:10.1021/acs.orglett.8b03843
Return to citation in text: [1] -
Li, L.; Guo, S.; Wang, Q.; Zhu, J. Org. Lett. 2019, 21, 5462–5466. doi:10.1021/acs.orglett.9b01717
Return to citation in text: [1] -
Abazid, A. H.; Clamor, N.; Nachtsheim, B. J. ACS Catal. 2020, 10, 8042–8048. doi:10.1021/acscatal.0c02321
Return to citation in text: [1] -
Abazid, A. H.; Nachtsheim, B. J. Angew. Chem., Int. Ed. 2020, 59, 1479–1484. doi:10.1002/anie.201912023
Return to citation in text: [1] -
Massignan, L.; Tan, X.; Meyer, T. H.; Kuniyil, R.; Messinis, A. M.; Ackermann, L. Angew. Chem., Int. Ed. 2020, 59, 3184–3189. doi:10.1002/anie.201914226
Return to citation in text: [1] -
Ghosh, S.; Pradhan, S.; Chatterjee, I. Beilstein J. Org. Chem. 2018, 14, 1244–1262. doi:10.3762/bjoc.14.107
Return to citation in text: [1] -
Jacquemot, G.; Maertens, G.; Canesi, S. Chem. – Eur. J. 2015, 21, 7713–7715. doi:10.1002/chem.201500185
Return to citation in text: [1] -
Maertens, G.; L'Homme, C.; Canesi, S. Front. Chem. (Lausanne, Switz.) 2015, 2. doi:10.3389/fchem.2014.00115
Return to citation in text: [1] -
Silva, L. F., Jr.; Olofsson, B. Nat. Prod. Rep. 2011, 28, 1722–1754. doi:10.1039/c1np00028d
Return to citation in text: [1] -
Ciufolini, M.; Braun, N.; Canesi, S.; Ousmer, M.; Chang, J.; Chai, D. Synthesis 2007, 3759–3772. doi:10.1055/s-2007-990906
Return to citation in text: [1] -
Hamamoto, H.; Shiozaki, Y.; Nambu, H.; Hata, K.; Tohma, H.; Kita, Y. Chem. – Eur. J. 2004, 10, 4977–4982. doi:10.1002/chem.200400358
Return to citation in text: [1] -
Novák, Z.; Aradi, K.; Tóth, B.; Tolnai, G. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] -
Chatterjee, N.; Goswami, A. Eur. J. Org. Chem. 2017, 3023–3032. doi:10.1002/ejoc.201601651
Return to citation in text: [1] -
Grushin, V. V. Chem. Soc. Rev. 2000, 29, 315–324. doi:10.1039/a909041j
Return to citation in text: [1] -
Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373
Return to citation in text: [1] -
Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974
Return to citation in text: [1] -
Caspers, L. D.; Spils, J.; Damrath, M.; Lork, E.; Nachtsheim, B. J. J. Org. Chem. 2020, 85, 9161–9178. doi:10.1021/acs.joc.0c01125
Return to citation in text: [1] [2] -
Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691
Return to citation in text: [1] -
Elsherbini, M.; Wirth, T. Chem. – Eur. J. 2018, 24, 13399–13407. doi:10.1002/chem.201801232
Return to citation in text: [1] -
Francke, R. Curr. Opin. Electrochem. 2019, 15, 83–88. doi:10.1016/j.coelec.2019.03.012
Return to citation in text: [1] -
Doobary, S.; Sedikides, A. T.; Caldora, H. P.; Poole, D. L.; Lennox, A. J. J. Angew. Chem., Int. Ed. 2020, 59, 1155–1160. doi:10.1002/anie.201912119
Return to citation in text: [1] -
Elsherbini, M.; Winterson, B.; Alharbi, H.; Folgueiras‐Amador, A. A.; Génot, C.; Wirth, T. Angew. Chem., Int. Ed. 2019, 58, 9811–9815. doi:10.1002/anie.201904379
Return to citation in text: [1] -
Maity, A.; Frey, B. L.; Hoskinson, N. D.; Powers, D. C. J. Am. Chem. Soc. 2020, 142, 4990–4995. doi:10.1021/jacs.9b13918
Return to citation in text: [1] -
Wirth, T. Curr. Opin. Electrochem. 2021, 28, 100701. doi:10.1016/j.coelec.2021.100701
Return to citation in text: [1] -
Winterson, B.; Rennigholtz, T.; Wirth, T. Chem. Sci. 2021, 12, 9053–9059. doi:10.1039/d1sc02123k
Return to citation in text: [1] -
Zu, B.; Ke, J.; Guo, Y.; He, C. Chin. J. Chem. 2021, 39, 627–632. doi:10.1002/cjoc.202000501
Return to citation in text: [1] [2] -
Elsherbini, M.; Moran, W. J. Org. Biomol. Chem. 2021, 19, 4706–4711. doi:10.1039/d1ob00457c
Return to citation in text: [1] [2] -
Watts, K.; Gattrell, W.; Wirth, T. Beilstein J. Org. Chem. 2011, 7, 1108–1114. doi:10.3762/bjoc.7.127
Return to citation in text: [1] [2] -
Peacock, M. J.; Pletcher, D. J. Electrochem. Soc. 2001, 148, D37. doi:10.1149/1.1353574
Return to citation in text: [1] -
Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497
Return to citation in text: [1] -
Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360
Return to citation in text: [1] -
Dohi, T.; Yamaoka, N.; Kita, Y. Tetrahedron 2010, 66, 5775–5785. doi:10.1016/j.tet.2010.04.116
Return to citation in text: [1] -
Eberson, L.; Hartshorn, M. P.; Persson, O.; Radner, F. Chem. Commun. 1996, 2105–2112. doi:10.1039/cc9960002105
Return to citation in text: [1]
| 1. | Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h |
| 2. | Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073 |
| 3. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 4. | Caspers, L. D.; Nachtsheim, B. J. Chem. – Asian J. 2018, 13, 1231–1247. doi:10.1002/asia.201800102 |
| 24. | Chatterjee, N.; Goswami, A. Eur. J. Org. Chem. 2017, 3023–3032. doi:10.1002/ejoc.201601651 |
| 25. | Grushin, V. V. Chem. Soc. Rev. 2000, 29, 315–324. doi:10.1039/a909041j |
| 28. | Caspers, L. D.; Spils, J.; Damrath, M.; Lork, E.; Nachtsheim, B. J. J. Org. Chem. 2020, 85, 9161–9178. doi:10.1021/acs.joc.0c01125 |
| 22. | Novák, Z.; Aradi, K.; Tóth, B.; Tolnai, G. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 23. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 16. | Ghosh, S.; Pradhan, S.; Chatterjee, I. Beilstein J. Org. Chem. 2018, 14, 1244–1262. doi:10.3762/bjoc.14.107 |
| 17. | Jacquemot, G.; Maertens, G.; Canesi, S. Chem. – Eur. J. 2015, 21, 7713–7715. doi:10.1002/chem.201500185 |
| 18. | Maertens, G.; L'Homme, C.; Canesi, S. Front. Chem. (Lausanne, Switz.) 2015, 2. doi:10.3389/fchem.2014.00115 |
| 19. | Silva, L. F., Jr.; Olofsson, B. Nat. Prod. Rep. 2011, 28, 1722–1754. doi:10.1039/c1np00028d |
| 20. | Ciufolini, M.; Braun, N.; Canesi, S.; Ousmer, M.; Chang, J.; Chai, D. Synthesis 2007, 3759–3772. doi:10.1055/s-2007-990906 |
| 21. | Hamamoto, H.; Shiozaki, Y.; Nambu, H.; Hata, K.; Tohma, H.; Kita, Y. Chem. – Eur. J. 2004, 10, 4977–4982. doi:10.1002/chem.200400358 |
| 37. | Zu, B.; Ke, J.; Guo, Y.; He, C. Chin. J. Chem. 2021, 39, 627–632. doi:10.1002/cjoc.202000501 |
| 38. | Elsherbini, M.; Moran, W. J. Org. Biomol. Chem. 2021, 19, 4706–4711. doi:10.1039/d1ob00457c |
| 5. | Baralle, A.; Fensterbank, L.; Goddard, J.-P.; Ollivier, C. Chem. – Eur. J. 2013, 19, 10809–10813. doi:10.1002/chem.201301449 |
| 6. | Flores, A.; Cots, E.; Bergès, J.; Muñiz, K. Adv. Synth. Catal. 2019, 361, 2–25. doi:10.1002/adsc.201800521 |
| 7. | Le Vaillant, F.; Garreau, M.; Nicolai, S.; Gryn'ova, G.; Corminboeuf, C.; Waser, J. Chem. Sci. 2018, 9, 5883–5889. doi:10.1039/c8sc01818a |
| 8. | Wang, D.; Mao, J.; Zhu, C. Chem. Sci. 2018, 9, 5805–5809. doi:10.1039/c8sc01763h |
| 9. | Boelke, A.; Nachtsheim, B. J. Adv. Synth. Catal. 2019, 362, 184–191. doi:10.1002/adsc.201901356 |
| 10. | Cots, E.; Flores, A.; Romero, R. M.; Muñiz, K. ChemSusChem 2019, 12, 3028–3031. doi:10.1002/cssc.201900360 |
| 11. | Declas, N.; Le Vaillant, F.; Waser, J. Org. Lett. 2019, 21, 524–528. doi:10.1021/acs.orglett.8b03843 |
| 12. | Li, L.; Guo, S.; Wang, Q.; Zhu, J. Org. Lett. 2019, 21, 5462–5466. doi:10.1021/acs.orglett.9b01717 |
| 13. | Abazid, A. H.; Clamor, N.; Nachtsheim, B. J. ACS Catal. 2020, 10, 8042–8048. doi:10.1021/acscatal.0c02321 |
| 14. | Abazid, A. H.; Nachtsheim, B. J. Angew. Chem., Int. Ed. 2020, 59, 1479–1484. doi:10.1002/anie.201912023 |
| 15. | Massignan, L.; Tan, X.; Meyer, T. H.; Kuniyil, R.; Messinis, A. M.; Ackermann, L. Angew. Chem., Int. Ed. 2020, 59, 3184–3189. doi:10.1002/anie.201914226 |
| 43. | Dohi, T.; Yamaoka, N.; Kita, Y. Tetrahedron 2010, 66, 5775–5785. doi:10.1016/j.tet.2010.04.116 |
| 44. | Eberson, L.; Hartshorn, M. P.; Persson, O.; Radner, F. Chem. Commun. 1996, 2105–2112. doi:10.1039/cc9960002105 |
| 31. | Francke, R. Curr. Opin. Electrochem. 2019, 15, 83–88. doi:10.1016/j.coelec.2019.03.012 |
| 32. | Doobary, S.; Sedikides, A. T.; Caldora, H. P.; Poole, D. L.; Lennox, A. J. J. Angew. Chem., Int. Ed. 2020, 59, 1155–1160. doi:10.1002/anie.201912119 |
| 33. | Elsherbini, M.; Winterson, B.; Alharbi, H.; Folgueiras‐Amador, A. A.; Génot, C.; Wirth, T. Angew. Chem., Int. Ed. 2019, 58, 9811–9815. doi:10.1002/anie.201904379 |
| 34. | Maity, A.; Frey, B. L.; Hoskinson, N. D.; Powers, D. C. J. Am. Chem. Soc. 2020, 142, 4990–4995. doi:10.1021/jacs.9b13918 |
| 35. | Wirth, T. Curr. Opin. Electrochem. 2021, 28, 100701. doi:10.1016/j.coelec.2021.100701 |
| 36. | Winterson, B.; Rennigholtz, T.; Wirth, T. Chem. Sci. 2021, 12, 9053–9059. doi:10.1039/d1sc02123k |
| 41. | Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497 |
| 42. | Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360 |
| 30. | Elsherbini, M.; Wirth, T. Chem. – Eur. J. 2018, 24, 13399–13407. doi:10.1002/chem.201801232 |
| 39. | Watts, K.; Gattrell, W.; Wirth, T. Beilstein J. Org. Chem. 2011, 7, 1108–1114. doi:10.3762/bjoc.7.127 |
| 28. | Caspers, L. D.; Spils, J.; Damrath, M.; Lork, E.; Nachtsheim, B. J. J. Org. Chem. 2020, 85, 9161–9178. doi:10.1021/acs.joc.0c01125 |
| 29. | Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691 |
| 26. | Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373 |
| 27. | Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974 |
| 37. | Zu, B.; Ke, J.; Guo, Y.; He, C. Chin. J. Chem. 2021, 39, 627–632. doi:10.1002/cjoc.202000501 |
| 38. | Elsherbini, M.; Moran, W. J. Org. Biomol. Chem. 2021, 19, 4706–4711. doi:10.1039/d1ob00457c |
| 39. | Watts, K.; Gattrell, W.; Wirth, T. Beilstein J. Org. Chem. 2011, 7, 1108–1114. doi:10.3762/bjoc.7.127 |
| 40. | Peacock, M. J.; Pletcher, D. J. Electrochem. Soc. 2001, 148, D37. doi:10.1149/1.1353574 |
© 2023 Spils et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.