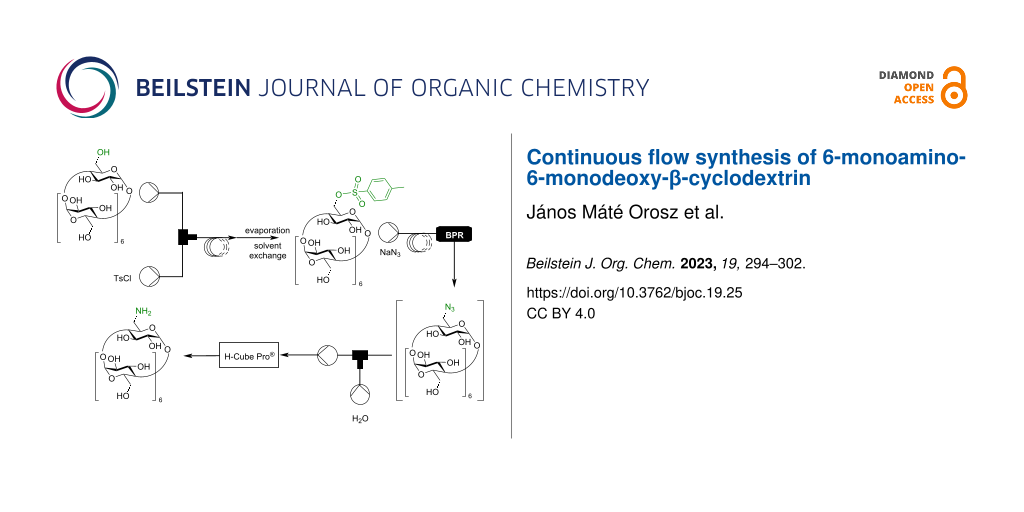
Abstract
The first continuous flow method was developed for the synthesis of 6-monoamino-6-monodeoxy-β-cyclodextrin starting from native β-cyclodextrin through three reaction steps, such as monotosylation, azidation and reduction. All reaction steps were studied separately and optimized under continuous flow conditions. After the optimization, the reaction steps were coupled in a semi-continuous flow system, since a solvent exchange had to be performed after the tosylation. However, the azidation and the reduction steps were compatible to be coupled in one flow system obtaining 6-monoamino-6-monodeoxy-β-cyclodextrin in a high yield. Our flow method developed is safer and faster than the batch approaches.

Graphical Abstract
Introduction
Cyclodextrins (CDs), as cyclic oligosaccharides, consist of a macrocyclic ring of glucose subunits linked by α-1,4-glycosidic bonds [1]. They are widely used in pharmaceutical, food and chemical industries, as well as in agriculture. It has been known for decades, that CD complexation can lead to a significant solubility enhancement of poorly water-soluble molecules, and therefore it can enable the biological testing of drugs, which would otherwise not be possible by any other means [2,3].
Monosubstituted CDs contain only one hydroxy group modified with a functional group. In most cases, the preparation of these compounds is based on the use of a limited amount of the reagent. However, due to the very similar reactivity of hydroxy groups, oversubstitution cannot be avoided during the reaction, thus chromatography or crystallization steps are essential for the preparation of pure monofunctionalized CDs. Alternative approaches use sterically hindered reagents, preventing the approach of the second molecule of the reagent to provide higher yields for the monosubstituted compounds [4]. The three different hydroxy groups on the glucose subunits offer three different sites on the CD molecule where the monofunctionalization can occur. Consequently, monosubstituted CDs can be mixtures of three regioisomers.
The number of known monosubstituted CD derivatives is enormous, since monosubstitution was the almost exclusive reaction for the practical production of selectively modified CDs for a long time. From the synthetic point of view, the most important derivatives are those versatile intermediates that can be effectively transformed according to the requirements of the specific application. The modification of a monosubstituted CD with a suitable functional group is an easier process than the optimization of a new monosubstitution reaction on a native CD [5].
Monotosylation of the primary rim of CDs is the most widely used method to obtain C-6 monofunctionalized CDs. Tosyl chloride (TsCl) reacts with α-, β-, and γ-CD in pyridine to give the C-6-monosubstituted product in about 30% yield (for β-CD) [6,7]. The C-6 regioselectivity can be attributed to the inclusion of pyridine into the CD cavity in such a way that it activates only the hydroxy groups on the primary side. Several alternative methods have been developed with the aim of further improving the yield of monotosylation or replacing pyridine with a more user-friendly solvent [8]. Regardless of which strategy is used, the complete conversion of the starting material into the monosubstituted product does not occur, and a mixture of overtosylated products and unreacted starting CD is formed. The target monosulfonated compound is separated by recrystallization from hot water in the case of the β-CD derivative, and by chromatography in the case of α- and γ-CDs.
The larger cavity size of γ-CD is the reason of polysubstitution when TsCl is used [9]. To ensure a better yield of the monosubstituted product, bulkier sulfonyl chloride reagents are used. 2-Naphthalenesulfonyl chloride in pyridine is one common method. However, the concentration of γ-CD must be lower than 20 mM to favor monosubstitution and to ensure the optimal yield (around 30%) after recrystallization from hot water [9]. Sometimes purification using ion exchange column [10] or reversed-phase chromatography has been reported [11]. An even bulkier sulfonyl chloride reagent, specifically 2,4,6-triisopropylbenzenesulfonyl chloride, can also be used [12]. Again, the reaction is performed in pyridine and the desired monosubstituted product is obtained in 69% yield and 98% purity according to the authors.
Many nucleophiles can react with tosylated CDs to give the corresponding C-6-monofunctionalized CDs. However, alkaline bases cannot be used as nucleophiles due to the intramolecular substitution, resulting in a mono-3,6-anhydro product [13]. On the other hand, sodium azide in N,N-dimethylformamide (DMF) reacts with mono-6-O-tosyl-CDs to give CD monoazides in high yields. The obtained mono(6-azido-6-deoxy)-CDs (N3-CDs) are valuable precursors that can be used as starting materials in azide–alkyne click reactions; furthermore, they can be readily reduced to mono(6-amino-6-deoxy)-CDs (NH2-CDs) opening the way for the preparation of amine, thioureido or amide-linked CD scaffolds [14]. Several other nucleophiles can react with mono-6-O-tosyl-CDs, such as iodide, dithiol, hydroxylamine, alkylamine or polyalkylamine to give iodo- [15], thio- [16], hydroxylamino- [17], or alkylamino-CDs [18], monosubstituted at position C-6. In addition, the tosyl functional group can be oxidized to an aldehyde using a non-nucleophilic base in dimethyl sulfoxide (DMSO) [19]. The monoaldehyde CDs can be further oxidized selectively to afford the corresponding carboxylic acid derivatives [20].
An alternative strategy to overcome the difficulties associated with the preparation of mono-6-O-tosyl-CD intermediates is the direct preparation of 6-monoaldehyde-CD with Dess–Martin periodinane in a fairly good yield of 85%, which can be considered the most efficient reaction used so far for the selective monofunctionalization of CDs [1].
Besides traditional synthetic methods, alternative techniques, such as ultrasound and microwave irradiation [21], as well as mechanosynthesis [22,23] for the functionalizations, such as tosylation or azidation of CDs have been also described [24,25].
Continuous flow approaches have already attracted much attention in the oil, plastic, and fine chemical industries [26]. The vision of faster, safer, cheaper, more flexible and robust production also initiated the paradigm shift from batch reactions to continuous flow processes in the pharmaceutical industry [1,27]. During continuous flow reactions, the target molecules can be produced with better purity, selectivity and in higher yields, as well as in consistent quality due to the precise parameter control, low volume ratio and small quantities. The temperature control is simple and toxic or unstable intermediates are easier to handle, making the overall process safer.
To the best of our knowledge, there is no publication on the continuous flow monosubstitution of CD derivatives. In this paper, first we wished to summarize the batch synthesis of 6A-O-(p-toluenesulfonyl)-β-CD, 6A-azido-6A-deoxy-β-CD, and 6A-amino-6A-deoxy-β-CD to see how compatible batch methods are with flow synthesis, and then our main aim was to develop continuous flow approaches for the preparation of the mentioned CD derivatives.
Results and Discussion
Batch synthesis of 6A-O-(p-toluenesulfonyl)-β-CD (Ts-β-CD, 2)
There are three standard methods for the preparation of this general and functional CD derivative. Two of them take advantage of the lower aqueous solubility of β-CD (1) and Ts-β-CD (2) compared to α- and γ-CD analogs. The third method is mainly used for Ts-α-CD and Ts-γ-CD synthesis, but Ts-β-CD (2) can also be prepared by this route. However, before discussing these methods in more detail, some issues related to tosylation reagents should be considered.
There are three tosylating agents utilized for the synthesis of Ts-β-CD, p-toluenesulfonyl chloride (TsCl) [14,28,29], (p-toluenesulfonyl)imidazole (TsIm) [30,31], and p-toluenesulfonic anhydride (Ts2O) [32]. TsCl is the first choice due to its low price and availability, being a byproduct of saccharin production. The two other reagents are more expensive or can be prepared in the laboratory from TsCl. No significant differences are observed in the product yields concerning the types of the tosylating agents.
As already mentioned above, there are three standard methods for the preparation of Ts-β-CD (2). The first and most common method can be called heterogeneous approach [28,33,34]. Here, solid TsCl (sufficiently crushed) is added to a β-CD (1) aqueous solution and this heterogeneous mixture is stirred for several hours. Then, aqueous NaOH solution is added and the mixture is stirred for another 10–20 minutes. Unreacted TsCl is filtered off and Ts-β-CD, overreacted byproducts, and β-CD are precipitated after neutralization.
The second method could bear the name homogeneous approach [8,14,35]. It is very similar to the previous method, however, TsCl dissolved in MeCN is added to the basic β-CD aqueous solution. Turbidity or even slight precipitation is observed during the process. After a few hours, the reaction mixture is filtered and neutralized to induce precipitation.
The third method works with pyridine as a solvent [29,36,37]. In this method, β-CD is dissolved in pyridine and a pyridine solution of TsCl is added dropwise. After a few hours, the solvent is distilled off and the residue is precipitated in acetone in order to obtain solid β-CD compounds.
Two important things need to be mentioned before closing this part. First, the amounts of TsCl range from 0.5 to 9 equiv in the literature and there is no direct correlation between the yield and the amount of TsCl. Usually 1 to 1.3 equiv are sufficient to ensure good yields. However, the yields bring us to the second important point. As already mentioned, the yield of this reaction is not strongly influenced by the amount of TsCl, but by the purification method. If only the precipitation is carried out, then the crude product is always a mixture of Ts-β-CD (2), overreacted byproducts, and starting β-CD (1). However, in previous studies this has been reported as a clean product with a yield of almost 50% [31,33]. Recrystallization is mandatory to obtain pure Ts-β-CD (2) [38,39]. The best results are obtained by crystallization of the crude product from 50% MeOH/water [18], which we adopted for our batch synthesis of Ts-β-CD (2). After proper purification, the yield of the desired product 2 was around 25%. Readers can also find more information on problematic p-toluenesulfonylation, subsequent azidation and reduction, in a recently published review [5].
It is clear from the already mentioned facts about the synthesis of Ts-β-CD (2), that neither of these methods is suitable for the flow chemistry process. Heterogeneous mixtures should be strictly avoided and pyridine is a toxic compound and should not be used in large-scale syntheses or industrial processes.
Batch synthesis of 6A-azido-6A-deoxy-β-CD (N3-β-CD) (3)
Substitution of the p-toluenesulfonyl group of Ts-β-CD (2) by azide can be carried out in water [40,41], DMF [14,42], or in their combination [43,44] at elevated temperatures. Water is preferred over DMF due to its lower cost and non-toxicity. However, partial hydrolysis of the p-toluenesulfonyl group takes place, so the final product is always contaminated with native β-CD (1). Despite this, the product mixture after precipitation from acetone was used in the next reaction step and a proper purification of the targeted monosubsituted compound was performed after this last modification. After precipitation, an apparent yield of more than 90% was noted; however, this value did not take into account native β-CD (1) as byproduct. On the other hand, if purification by column chromatography or crystallization is also used, yields of 60–80% can be achieved.
Partial hydrolysis can be avoided by using anhydrous DMF and purifying the starting Ts-β-CD (2) by crystallization. In this case, a lower amount of NaN3 (1–2 equiv) is required and only purification by precipitation from acetone is necessary. In order to prepare N3-β-CD (3) in batch, we decided to follow the protocol developed by Jicsinszky and Iványi [14], who performed this reaction in anhydrous DMF using 1.1 equiv of NaN3 at 110 °C. The product 3 was purified by repeated precipitation from acetone and isolated in a yield of 81%.
Batch synthesis of 6A-amino-6A-deoxy-β-CD (NH2-β-CD) (4)
Again, there are several common methods for the preparation of NH2-β-CD (4). The most straightforward route is the reaction of Ts-β-CD (2) with condensed NH3 in dry DMF [45]. However, this reaction leads to a complex mixture and the yield is around 50% after proper purification.
The Staudinger reduction using triphenylphosphine (PPh3) and N3-β-CD (3) in DMF has been the most popular method for the synthesis of NH2-β-CD (4) since its first publication by Bonnet et al. [46]. This is despite the fact, that PPh3 and its oxidized product (triphenylphosphine oxide) form complexes with β-CD derivatives. This complexation creates difficulties in the purification process, which consists mostly of precipitation from acetone or ion-exchange column chromatography. The price of the reagent needs to be also considered in the case of an attempted large-scale synthesis.
The second most used method is the hydrogenation of N3-β-CD (3) in the presence of Pd/C under a H2 atmosphere. In CD chemistry, this method was first described by Petter et al. [8] in the early 1990s. This method is very popular with small-scale syntheses because only gaseous N2 is formed as a byproduct and no purification is required when pure N3-β-CD (3) is used as substrate. However, for large-scale syntheses, mixing hydrogen with the Pd/C catalyst can be dangerous if an inert atmosphere is not properly maintained. In addition, Pd/C is pyrophoric and tends to ignite when it is separated from the reaction mixture by filtration. However, these two drawbacks are not a problem for large-scale flow synthesis when using an H-Cube system with an incorporated electrolytic cell producing H2 in situ from ultrapure water [47]. The Pd/C catalyst is placed in a stainless steel cartridge, so it is not necessary to separate it from the solution after the reaction is complete.
Hydrazine hydrate can also be used as a hydrogen source instead of gaseous H2, as described by Jicsinszky and Iványi [14], although this method is not so widespread and has limited potential for a large-scale synthesis. As a special feature, the protocol published by Reddy et al. [48] is worth mentioning, who used metallic indium and ammonium chloride for the reduction of N3-β-CD.
Continuous flow synthesis of 6A-O-(p-toluenesulfonyl)-β-CD (2)
According to the literature, most of the tosylations of CDs took place under heterogeneous conditions. However, small solid particles can easily cause clogging in the narrow tubing of a flow system, therefore homogenous solutions were tried to introduce into the flow systems in all experiments. Regarding the tosylation, β-CD (1) and the required base (NaOH) are soluble in water, however, TsCl needs to be dissolved in organic solvents. Alcohols were excluded as possible solvents, as they precipitate β-CD (1) and may cause side reactions, but aprotic solvents such as tetrahydrofuran (THF) or acetonitrile (MeCN) were found to be suitable for homogenous conditions, especially a H2O/THF 2:1 mixture. This solvent mixture was prepared in situ in the flow tube reactor, as the aqueous solution containing β-CD (1) and NaOH (1.5 equiv) was introduced into the reactor at twice the flow rate as the solution of TsCl in THF (Scheme 1).
![[1860-5397-19-25-i1]](/bjoc/content/inline/1860-5397-19-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Tosylation of β-CD under continuous flow conditions.
Scheme 1: Tosylation of β-CD under continuous flow conditions.
In this way, twice as many equivalents of TsCl were required compared to the general 1.3 equivalents used in the batch process. The reaction residence time (τ) was varied between 2.2 and 5.3 minutes (Table 1, entries 1–4). The 3 minute residence time was found to be the optimal one and led to a conversion of 24% (Table 1, entry 2). This is a significant advantage compared to batch methods, where similar conversions were achieved in over 2 hours. The flow tosylation was also investigated using acetonitrile instead of THF, however, a much lower conversion was observed (Table 1, entry 4).
Table 1: Optimization of the tosylation of β-CD (1) under continuous flow conditions.
| Entry | Flow rate of TsCl solution [mL/min] |
Flow rate of β-CD solution
[mL/min] |
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-25-i5.svg?max-width=637&scale=1.0)
[min] |
Solvent |
Conversiona
[%] |
| 1 | 0.60 | 1.20 | 2.2 | H2O/THF 2:1 | 22 |
| 2 | 0.45 | 0.90 | 3.0 | H2O/THF 2:1 | 24 |
| 3 | 0.25 | 0.50 | 5.3 | H2O/THF 2:1 | 17 |
| 4 | 0.25 | 0.50 | 5.3 | H2O/MeCN 2:1 | 11 |
aOn the basis of HPLC (214 nm).
Continuous flow synthesis of 6A-azido-6A-deoxy-β-CD (3)
After the tosylation step was successful in flow, next the tosyl–azide substitution was optimized (Scheme 2).
![[1860-5397-19-25-i2]](/bjoc/content/inline/1860-5397-19-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Continuous flow azidation of Ts-β-CD (2).
Scheme 2: Continuous flow azidation of Ts-β-CD (2).
First, the best solvent was sought. Unfortunately, practically no reaction took place when the same solvent was used as for the tosylation reaction (Table 2, entries 1–4), so we had to evaporate the solution exiting the first flow reaction, and change the solvent. According to the literature, DMF proved to be a suitable solvent to conduct this reaction under homogenous conditions. By prolonging the residence time, the conversion greatly increased (Table 2, entries 5–7). However, with longer residence times, increasing hydrolysis of Ts-β-CD (2) was observed. To solve this issue, water was excluded from the azidation, which resulted in a total conversion to the corresponding N3-β-CD (3) (Table 2, entries 8–10). In order allow comparison of the results with those from the batch synthesis, the amount of NaN3 was decreased to 1.1 equivalents (Table 2, entries 13–16). Ultimately, the reactor was heated to its limit at 125 °C, by which a satisfying conversion could be achieved. Thus, it can be concluded, that a similar conversion in flow as compared to the batch azidation could be obtained and, in addition as a major advantage, the reaction time was greatly reduced to only 10 minutes.
Table 2: Optimization of the continuous flow azidation of Ts-β-CD (2).
| Entry |
NaN3
[equiv] |
Solvent |
T
[°C] |
Total flow rate
[mL/min] |
[min] |
Conversiona
[%] |
| 1b | 2 | H2O/THF 2:1 | 25 | 1.5 | 2.7 | 0 |
| 2c | 2 | H2O/THF 2:1 | 25 | 1.5 | 2.7 | 0 |
| 3 | 2 | H2O/THF 2:1 | 110 | 1.5 | 2.7 | 0 |
| 4 | 8 | H2O/THF 2:1 | 110 | 1.5 | 2.7 | 11 |
| 5 | 8 | H2O/DMF 1:1 | 110 | 0.75 | 5.3 | 39 |
| 6 | 8 | H2O/DMF 1:1 | 110 | 0.40 | 10 | 45 |
| 7 | 8 | H2O/DMF 1:1 | 110 | 0.20 | 20 | 60 |
| 8 | 8 | DMF | 110 | 0.40 | 10 | 100 |
| 9 | 6 | DMF | 110 | 0.40 | 10 | 100 |
| 10 | 4 | DMF | 110 | 0.40 | 10 | 100 |
| 11 | 3 | DMF | 110 | 0.40 | 10 | 96 |
| 12 | 2 | DMF | 110 | 0.40 | 10 | 83 |
| 13 | 1.1 | DMF | 110 | 0.40 | 10 | 45 |
| 14 | 1.1 | DMF | 125 | 0.40 | 10 | 86 |
| 15 | 1.1 | DMF | 125 | 0.20 | 20 | 85 |
| 16 | 1.1 | DMF | 125 | 0.13 | 30 | 78 |
aBased on HPLC (214 nm). bThe reaction was carried out using the reaction mixture obtained by the flow tosylation. cPure starting material was used, which was obtained from batch tosylation.
Continuous flow synthesis of 6A-amino-6A-deoxy-β-CD (4)
In the last step of the flow synthesis, the reduction of the N3-β-CD (3) was investigated in an H-Cube Pro® flow hydrogenating reactor containing a 10% Pd/C pre-packed cartridge (Scheme 3). Generally, a 1 mL/min input flow was used while conducting the hydrogenations during the optimization, which resulted in approximately a 20 second residence time in all cases.
![[1860-5397-19-25-i3]](/bjoc/content/inline/1860-5397-19-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Continuous flow hydrogenation of N3-β-CD (3).
Scheme 3: Continuous flow hydrogenation of N3-β-CD (3).
A complete conversion was observed using even the mildest conditions possible (25 °C, 1 bar H2 pressure) in aqueous solution (Table 3, entry 1). In spite of this excellent result, the previous azidation reaction proceeded well in DMF, so in order to connect the single flow steps together, the effect of DMF and water/DMF mixtures as solvents had to be studied as well. In DMF, the reaction was not complete and only 30% of NH2-β-CD (4) were obtained (Table 3, entry 2). However, increasing the ratio of water in the solvent mixture afforded better results (Table 3, entries 3–6). The appropriate ratio of DMF/H2O was determined as 1:4, which gave NH2-β-CD (4) in a yield of 93% (Table 3, entry 5).
Table 3: Optimization of the continuous flow hydrogenation of N3-β-CD (3).
| Entry | Solvent | Yielda [%] |
| 1 | H2O | 94 |
| 2 | DMF | 30 |
| 3 | DMF/H2O 1:1 | 51 |
| 4 | DMF/H2O 1:3 | 85 |
| 5 | DMF/H2O 1:4 | 93 |
| 6 | DMF/H2O 1:9 | 94 |
aIsolated yield.
Semi-continuous flow system for the synthesis of 6A-amino-6A-deoxy-β-CD (4)
The main goal of this part of the work was to establish a continuous flow method for the synthesis of NH2-β-CD (4) from β-CD (1). Thus, after the optimization of the single reaction steps, the connection of these steps was remaining. First of all, it was concluded, that the initial tosylation step could not be connected to the azidation step since in the tosylation’s optimal solvent, the azidation did not proceed and vice versa (Table 4). The only solution was to conduct the tosylation under flow conditions separately from the other two steps (Scheme 4I), evaporate the water/THF solvent, and introduce the Ts-β-CD (2) dissolved in DMF to the second part of the flow system. In order to simplify the azidation and subsequent reduction, Ts-β-CD (2) prepared from batch and properly purified was utilized.
Table 4: Optimal reaction conditions for each flow reaction step.
| Synthesis step | Reagent | Solvent | T [°C] |
[min]
|
Conversiona [%] |
| tosylation | 2.6 equiv TsCl | H2O/THF 2:1 | 25 | 3 | 20 |
| azidation | 1.1 equiv NaN3 | DMF | 125 | 10 | 81 |
| hydrogenationb | H2 | H2O/DMF 1:4 | 25 | 0.3 | 93 |
aIsolated yield. b1 bar H2 pressure and 10 mol % Pd/C catalyst were used.
![[1860-5397-19-25-i4]](/bjoc/content/inline/1860-5397-19-25-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Semi-continuous flow system for the synthesis of NH2-β-CD 4.
Scheme 4: Semi-continuous flow system for the synthesis of NH2-β-CD 4.
The azidation and the hydrogenation were compatible with each other, however, after the azidation took place, water needed to be introduced to the system before the hydrogenation to ensure full conversion during the reduction. According to our previous results, the reduction of N3-β-CD (3) went to completion in a DMF/H2O 1:4 mixture, so this solvent was chosen for the hydrogenation step in order to avoid re-optimization. This way, the exiting solution of 0.4 mL/min from the azidation reaction was joined with a 1.6 mL/min flow of water, and this new solution was gathered in a buffer container, from where the reaction mixture was immediately forwarded to the H-Cube Pro® (Scheme 4II). Although the hydrogenation was optimized for a 1 mL/min input flow rate, the catalyst cartridge was changed to another one, which doubled the reaction volume. In conclusion, a 2 mL/min input flow rate required no re-optimization, as the residence time remained the same. Under these conditions, the connected azidation and reduction steps led to an almost complete conversion of Ts-β-CD (2). The output of the H-Cube Pro® was partially evaporated, the product 4 precipitated with acetone and isolated in a yield of 91%.
Conclusion
In conclusion, we have developed continuous flow methods for the monotosylation of β-CD (1), for the azidation of 6A-O-(p-toluenesulfonyl)-β-CD (2), and for the reduction of 6A-azido-6A-deoxy-β-CD (3). The flow methods are novel approaches for the preparation of the target compounds and were optimized for each case. Comparing the flow processes with batch methods, it can be concluded that similar yields were obtained in both cases, however, under continuous flow conditions, the reaction time could be reduced from hours to minutes. Finally, we made an attempt to connect the three reaction steps with each other in a continuous flow system. It was found that a solvent exchange step was required after the tosylation, however, the azidation and the reduction steps were compatible to be coupled in one flow system. Using our semi-flow method developed, the production of 6A-amino-6A-deoxy-β-CD (4) could be carried out in a safer way due to the easier handling of toxic derivatives and with more precise parameter control. Moreover, the reactions can be performed within a much shorter reaction time than under batch conditions.
Supporting Information
Experimental procedures, characterization data, details of the NMR structural determination of the products and copies of 1H NMR spectra for the compounds synthesized.
| Supporting Information File 1: Experimental section and copies of NMR spectra. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
The authors thank GB and PK for their contribution to the work with their Ph.D. dissertations.
Funding
This research was funded by the Hungarian Research Development and Innovation Office (FK142712) and by the Cylolab Grant. This work was performed in the frame of the Pharmaceutical Research and Development Laboratory project (PharmaLab, RRF-2.3.1-21-2022-00015), implemented with the support provided from the Széchenyi Plan Plus, financed under the National Laboratory Program funding scheme.
References
-
Szejtli, J. Chem. Rev. 1998, 98, 1743–1754. doi:10.1021/cr970022c
Return to citation in text: [1] [2] [3] -
Freudenberg, K.; Cramer, F.; Plieninger, H. Process for the preparation of inclusion compounds of physiologically active organic compounds. Ger. Patent DE895769C, May 11, 1953.
Return to citation in text: [1] -
Crini, G. Chem. Rev. 2014, 114, 10940–10975. doi:10.1021/cr500081p
Return to citation in text: [1] -
Cornwell, M. J.; Huff, J. B.; Bieniarz, C. Tetrahedron Lett. 1995, 36, 8371–8374. doi:10.1016/0040-4039(95)01808-u
Return to citation in text: [1] -
Kasal, P.; Jindřich, J. Molecules 2021, 26, 5065. doi:10.3390/molecules26165065
Return to citation in text: [1] [2] -
Melton, L. D.; Slessor, K. N. Carbohydr. Res. 1971, 18, 29–37. doi:10.1016/s0008-6215(00)80256-6
Return to citation in text: [1] -
Matsui, Y.; Yokoi, T.; Mochida, K. Chem. Lett. 1976, 5, 1037–1040. doi:10.1246/cl.1976.1037
Return to citation in text: [1] -
Petter, R. C.; Salek, J. S.; Sikorski, C. T.; Kumaravel, G.; Lin, F.-T. J. Am. Chem. Soc. 1990, 112, 3860–3868. doi:10.1021/ja00166a021
Return to citation in text: [1] [2] [3] -
Djedaïni-Pilard, F.; Azaroual-Bellanger, N.; Gosnat, M.; Vernet, D.; Perly, B. J. Chem. Soc., Perkin Trans. 2 1995, 723–730. doi:10.1039/p29950000723
Return to citation in text: [1] [2] -
Uekama, K.; Minami, K.; Hirayama, F. J. Med. Chem. 1997, 40, 2755–2761. doi:10.1021/jm970130r
Return to citation in text: [1] -
Kahee, F.; Tsutomu, T.; Taiji, I.; Toshitaka, K. Chem. Lett. 1988, 17, 1329–1332. doi:10.1246/cl.1988.1329
Return to citation in text: [1] -
Palin, R.; Grove, S. J. A.; Prosser, A. B.; Zhang, M.-Q. Tetrahedron Lett. 2001, 42, 8897–8899. doi:10.1016/s0040-4039(01)01934-7
Return to citation in text: [1] -
Fujita, K.; Yamamura, H.; Imoto, T.; Tabushi, I. Chem. Lett. 1988, 17, 543–546. doi:10.1246/cl.1988.543
Return to citation in text: [1] -
Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ueno, A.; Moriwaki, F.; Osa, T.; Hamada, F.; Murai, K. Tetrahedron 1987, 43, 1571–1578. doi:10.1016/s0040-4020(01)90271-6
Return to citation in text: [1] -
Bednářová, E.; Hybelbauerová, S.; Jindřich, J. Beilstein J. Org. Chem. 2016, 12, 349–352. doi:10.3762/bjoc.12.38
Return to citation in text: [1] -
Fikes, L. E.; Winn, D. T.; Sweger, R. W.; Johnson, M. P.; Czarnik, A. W. J. Am. Chem. Soc. 1992, 114, 1493–1495. doi:10.1021/ja00030a062
Return to citation in text: [1] -
Popr, M.; Hybelbauerová, S.; Jindřich, J. Beilstein J. Org. Chem. 2014, 10, 1390–1396. doi:10.3762/bjoc.10.142
Return to citation in text: [1] [2] -
Martin, K. A.; Czarnik, A. W. Tetrahedron Lett. 1994, 35, 6781–6782. doi:10.1016/0040-4039(94)85003-8
Return to citation in text: [1] -
Yoon, J.; Hong, S.; Martin, K. A.; Czarnik, A. W. J. Org. Chem. 1995, 60, 2792–2795. doi:10.1021/jo00114a030
Return to citation in text: [1] -
Martina, K.; Trotta, F.; Robaldo, B.; Belliardi, N.; Jicsinszky, L.; Cravotto, G. Tetrahedron Lett. 2007, 48, 9185–9189. doi:10.1016/j.tetlet.2007.10.104
Return to citation in text: [1] -
Menuel, S.; Saitzek, S.; Monflier, E.; Hapiot, F. Beilstein J. Org. Chem. 2020, 16, 2598–2606. doi:10.3762/bjoc.16.211
Return to citation in text: [1] -
Menuel, S.; Doumert, B.; Saitzek, S.; Ponchel, A.; Delevoye, L.; Monflier, E.; Hapiot, F. J. Org. Chem. 2015, 80, 6259–6266. doi:10.1021/acs.joc.5b00697
Return to citation in text: [1] -
Cravotto, G.; Caporaso, M.; Jicsinszky, L.; Martina, K. Beilstein J. Org. Chem. 2016, 12, 278–294. doi:10.3762/bjoc.12.30
Return to citation in text: [1] -
Jicsinszky, L.; Rossi, F.; Solarino, R.; Cravotto, G. Molecules 2023, 28, 467. doi:10.3390/molecules28020467
Return to citation in text: [1] -
Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Wiley-VCH: Weinheim, Germany, 2000.
Return to citation in text: [1] -
Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183
Return to citation in text: [1] -
Hacket, F.; Simova, S.; Schneider, H.-J. J. Phys. Org. Chem. 2001, 14, 159–170. doi:10.1002/poc.348
Return to citation in text: [1] [2] -
Defaye, J.; Gadelle, A.; Guiller, A.; Darcy, R.; O'Sullivan, T. Carbohydr. Res. 1989, 192, 251–258. doi:10.1016/0008-6215(89)85184-5
Return to citation in text: [1] [2] -
Byun, H.-S.; Zhong, N.; Bittman, R. Org. Synth. 2000, 77, 225. doi:10.15227/orgsyn.077.0225
Return to citation in text: [1] -
Trotta, F.; Martina, K.; Robaldo, B.; Barge, A.; Cravotto, G. J. Inclusion Phenom. Macrocyclic Chem. 2007, 57, 3–7. doi:10.1007/s10847-006-9169-z
Return to citation in text: [1] [2] -
Zhong, N.; Byun, H.-S.; Bittman, R. Tetrahedron Lett. 1998, 39, 2919–2920. doi:10.1016/s0040-4039(98)00417-1
Return to citation in text: [1] -
McNaughton, M.; Engman, L.; Birmingham, A.; Powis, G.; Cotgreave, I. A. J. Med. Chem. 2004, 47, 233–239. doi:10.1021/jm030916r
Return to citation in text: [1] [2] -
Tripodo, G.; Wischke, C.; Neffe, A. T.; Lendlein, A. Carbohydr. Res. 2013, 381, 59–63. doi:10.1016/j.carres.2013.08.018
Return to citation in text: [1] -
Vizitiu, D.; Walkinshaw, C. S.; Gorin, B. I.; Thatcher, G. R. J. J. Org. Chem. 1997, 62, 8760–8766. doi:10.1021/jo9711549
Return to citation in text: [1] -
Ekberg, B.; Andersson, L. I.; Mosbach, K. Carbohydr. Res. 1989, 192, 111–117. doi:10.1016/0008-6215(89)85171-7
Return to citation in text: [1] -
Sforza, S.; Galaverna, G.; Corradini, R.; Dossena, A.; Marchelli, R. J. Am. Soc. Mass Spectrom. 2003, 14, 124–135. doi:10.1016/s1044-0305(02)00853-x
Return to citation in text: [1] -
Brady, B.; Lynam, N.; O'Sullivan, T.; Ahern, C.; Darcy, R. Org. Synth. 2000, 77, 220. doi:10.15227/orgsyn.077.0220
Return to citation in text: [1] -
Xu, M.; Wu, S.; Zeng, F.; Yu, C. Langmuir 2010, 26, 4529–4534. doi:10.1021/la9033244
Return to citation in text: [1] -
Uchida, W.; Yoshikawa, M.; Seki, T.; Miki, R.; Seki, T.; Fujihara, T.; Ishimaru, Y.; Egawa, Y. J. Inclusion Phenom. Macrocyclic Chem. 2017, 89, 281–288. doi:10.1007/s10847-017-0755-z
Return to citation in text: [1] -
Quan, C.-Y.; Chen, J.-X.; Wang, H.-Y.; Li, C.; Chang, C.; Zhang, X.-Z.; Zhuo, R.-X. ACS Nano 2010, 4, 4211–4219. doi:10.1021/nn100534q
Return to citation in text: [1] -
Chwalek, M.; Auzély, R.; Fort, S. Org. Biomol. Chem. 2009, 7, 1680–1688. doi:10.1039/b822976g
Return to citation in text: [1] -
Yang, F.; Zhang, Y.; Guo, H. New J. Chem. 2013, 37, 2275–2279. doi:10.1039/c3nj00474k
Return to citation in text: [1] -
Xu, W.; Liang, W.; Wu, W.; Fan, C.; Rao, M.; Su, D.; Zhong, Z.; Yang, C. Chem. – Eur. J. 2018, 24, 16677–16685. doi:10.1002/chem.201804001
Return to citation in text: [1] -
Brown, S. E.; Coates, J. H.; Coghlan, D. R.; Easton, C. J.; Vaneyk, S. J.; Janowski, W.; Lepore, A.; Lincoln, S. F.; Luo, Y.; May, B. L.; Schiesser, D. S.; Wang, P.; Williams, M. L. Aust. J. Chem. 1993, 46, 953–958. doi:10.1071/ch9930953
Return to citation in text: [1] -
Bonnet, V.; Duval, R.; Tran, V.; Rabiller, C. Eur. J. Org. Chem. 2003, 4810–4818. doi:10.1002/ejoc.200300449
Return to citation in text: [1] -
Jones, R. V.; Godorhazy, L.; Varga, N.; Szalay, D.; Urge, L.; Darvas, F. J. Comb. Chem. 2006, 8, 110–116. doi:10.1021/cc050107o
Return to citation in text: [1] -
Reddy, L. R.; Reddy, M. A.; Bhanumathi, N.; Rao, K. R. Indian J. Chem., Sect. B 2002, 41B, 645–646.
Return to citation in text: [1]
| 32. | Zhong, N.; Byun, H.-S.; Bittman, R. Tetrahedron Lett. 1998, 39, 2919–2920. doi:10.1016/s0040-4039(98)00417-1 |
| 28. | Hacket, F.; Simova, S.; Schneider, H.-J. J. Phys. Org. Chem. 2001, 14, 159–170. doi:10.1002/poc.348 |
| 33. | McNaughton, M.; Engman, L.; Birmingham, A.; Powis, G.; Cotgreave, I. A. J. Med. Chem. 2004, 47, 233–239. doi:10.1021/jm030916r |
| 34. | Tripodo, G.; Wischke, C.; Neffe, A. T.; Lendlein, A. Carbohydr. Res. 2013, 381, 59–63. doi:10.1016/j.carres.2013.08.018 |
| 8. | Petter, R. C.; Salek, J. S.; Sikorski, C. T.; Kumaravel, G.; Lin, F.-T. J. Am. Chem. Soc. 1990, 112, 3860–3868. doi:10.1021/ja00166a021 |
| 14. | Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2 |
| 35. | Vizitiu, D.; Walkinshaw, C. S.; Gorin, B. I.; Thatcher, G. R. J. J. Org. Chem. 1997, 62, 8760–8766. doi:10.1021/jo9711549 |
| 6. | Melton, L. D.; Slessor, K. N. Carbohydr. Res. 1971, 18, 29–37. doi:10.1016/s0008-6215(00)80256-6 |
| 7. | Matsui, Y.; Yokoi, T.; Mochida, K. Chem. Lett. 1976, 5, 1037–1040. doi:10.1246/cl.1976.1037 |
| 16. | Bednářová, E.; Hybelbauerová, S.; Jindřich, J. Beilstein J. Org. Chem. 2016, 12, 349–352. doi:10.3762/bjoc.12.38 |
| 14. | Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2 |
| 42. | Chwalek, M.; Auzély, R.; Fort, S. Org. Biomol. Chem. 2009, 7, 1680–1688. doi:10.1039/b822976g |
| 5. | Kasal, P.; Jindřich, J. Molecules 2021, 26, 5065. doi:10.3390/molecules26165065 |
| 17. | Fikes, L. E.; Winn, D. T.; Sweger, R. W.; Johnson, M. P.; Czarnik, A. W. J. Am. Chem. Soc. 1992, 114, 1493–1495. doi:10.1021/ja00030a062 |
| 43. | Yang, F.; Zhang, Y.; Guo, H. New J. Chem. 2013, 37, 2275–2279. doi:10.1039/c3nj00474k |
| 44. | Xu, W.; Liang, W.; Wu, W.; Fan, C.; Rao, M.; Su, D.; Zhong, Z.; Yang, C. Chem. – Eur. J. 2018, 24, 16677–16685. doi:10.1002/chem.201804001 |
| 4. | Cornwell, M. J.; Huff, J. B.; Bieniarz, C. Tetrahedron Lett. 1995, 36, 8371–8374. doi:10.1016/0040-4039(95)01808-u |
| 14. | Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2 |
| 5. | Kasal, P.; Jindřich, J. Molecules 2021, 26, 5065. doi:10.3390/molecules26165065 |
| 2. | Freudenberg, K.; Cramer, F.; Plieninger, H. Process for the preparation of inclusion compounds of physiologically active organic compounds. Ger. Patent DE895769C, May 11, 1953. |
| 3. | Crini, G. Chem. Rev. 2014, 114, 10940–10975. doi:10.1021/cr500081p |
| 15. | Ueno, A.; Moriwaki, F.; Osa, T.; Hamada, F.; Murai, K. Tetrahedron 1987, 43, 1571–1578. doi:10.1016/s0040-4020(01)90271-6 |
| 40. | Uchida, W.; Yoshikawa, M.; Seki, T.; Miki, R.; Seki, T.; Fujihara, T.; Ishimaru, Y.; Egawa, Y. J. Inclusion Phenom. Macrocyclic Chem. 2017, 89, 281–288. doi:10.1007/s10847-017-0755-z |
| 41. | Quan, C.-Y.; Chen, J.-X.; Wang, H.-Y.; Li, C.; Chang, C.; Zhang, X.-Z.; Zhuo, R.-X. ACS Nano 2010, 4, 4211–4219. doi:10.1021/nn100534q |
| 10. | Uekama, K.; Minami, K.; Hirayama, F. J. Med. Chem. 1997, 40, 2755–2761. doi:10.1021/jm970130r |
| 12. | Palin, R.; Grove, S. J. A.; Prosser, A. B.; Zhang, M.-Q. Tetrahedron Lett. 2001, 42, 8897–8899. doi:10.1016/s0040-4039(01)01934-7 |
| 38. | Brady, B.; Lynam, N.; O'Sullivan, T.; Ahern, C.; Darcy, R. Org. Synth. 2000, 77, 220. doi:10.15227/orgsyn.077.0220 |
| 39. | Xu, M.; Wu, S.; Zeng, F.; Yu, C. Langmuir 2010, 26, 4529–4534. doi:10.1021/la9033244 |
| 9. | Djedaïni-Pilard, F.; Azaroual-Bellanger, N.; Gosnat, M.; Vernet, D.; Perly, B. J. Chem. Soc., Perkin Trans. 2 1995, 723–730. doi:10.1039/p29950000723 |
| 13. | Fujita, K.; Yamamura, H.; Imoto, T.; Tabushi, I. Chem. Lett. 1988, 17, 543–546. doi:10.1246/cl.1988.543 |
| 18. | Popr, M.; Hybelbauerová, S.; Jindřich, J. Beilstein J. Org. Chem. 2014, 10, 1390–1396. doi:10.3762/bjoc.10.142 |
| 9. | Djedaïni-Pilard, F.; Azaroual-Bellanger, N.; Gosnat, M.; Vernet, D.; Perly, B. J. Chem. Soc., Perkin Trans. 2 1995, 723–730. doi:10.1039/p29950000723 |
| 29. | Defaye, J.; Gadelle, A.; Guiller, A.; Darcy, R.; O'Sullivan, T. Carbohydr. Res. 1989, 192, 251–258. doi:10.1016/0008-6215(89)85184-5 |
| 36. | Ekberg, B.; Andersson, L. I.; Mosbach, K. Carbohydr. Res. 1989, 192, 111–117. doi:10.1016/0008-6215(89)85171-7 |
| 37. | Sforza, S.; Galaverna, G.; Corradini, R.; Dossena, A.; Marchelli, R. J. Am. Soc. Mass Spectrom. 2003, 14, 124–135. doi:10.1016/s1044-0305(02)00853-x |
| 8. | Petter, R. C.; Salek, J. S.; Sikorski, C. T.; Kumaravel, G.; Lin, F.-T. J. Am. Chem. Soc. 1990, 112, 3860–3868. doi:10.1021/ja00166a021 |
| 11. | Kahee, F.; Tsutomu, T.; Taiji, I.; Toshitaka, K. Chem. Lett. 1988, 17, 1329–1332. doi:10.1246/cl.1988.1329 |
| 31. | Trotta, F.; Martina, K.; Robaldo, B.; Barge, A.; Cravotto, G. J. Inclusion Phenom. Macrocyclic Chem. 2007, 57, 3–7. doi:10.1007/s10847-006-9169-z |
| 33. | McNaughton, M.; Engman, L.; Birmingham, A.; Powis, G.; Cotgreave, I. A. J. Med. Chem. 2004, 47, 233–239. doi:10.1021/jm030916r |
| 20. | Yoon, J.; Hong, S.; Martin, K. A.; Czarnik, A. W. J. Org. Chem. 1995, 60, 2792–2795. doi:10.1021/jo00114a030 |
| 18. | Popr, M.; Hybelbauerová, S.; Jindřich, J. Beilstein J. Org. Chem. 2014, 10, 1390–1396. doi:10.3762/bjoc.10.142 |
| 14. | Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2 |
| 19. | Martin, K. A.; Czarnik, A. W. Tetrahedron Lett. 1994, 35, 6781–6782. doi:10.1016/0040-4039(94)85003-8 |
| 45. | Brown, S. E.; Coates, J. H.; Coghlan, D. R.; Easton, C. J.; Vaneyk, S. J.; Janowski, W.; Lepore, A.; Lincoln, S. F.; Luo, Y.; May, B. L.; Schiesser, D. S.; Wang, P.; Williams, M. L. Aust. J. Chem. 1993, 46, 953–958. doi:10.1071/ch9930953 |
| 46. | Bonnet, V.; Duval, R.; Tran, V.; Rabiller, C. Eur. J. Org. Chem. 2003, 4810–4818. doi:10.1002/ejoc.200300449 |
| 14. | Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2 |
| 28. | Hacket, F.; Simova, S.; Schneider, H.-J. J. Phys. Org. Chem. 2001, 14, 159–170. doi:10.1002/poc.348 |
| 29. | Defaye, J.; Gadelle, A.; Guiller, A.; Darcy, R.; O'Sullivan, T. Carbohydr. Res. 1989, 192, 251–258. doi:10.1016/0008-6215(89)85184-5 |
| 30. | Byun, H.-S.; Zhong, N.; Bittman, R. Org. Synth. 2000, 77, 225. doi:10.15227/orgsyn.077.0225 |
| 31. | Trotta, F.; Martina, K.; Robaldo, B.; Barge, A.; Cravotto, G. J. Inclusion Phenom. Macrocyclic Chem. 2007, 57, 3–7. doi:10.1007/s10847-006-9169-z |
| 26. | Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Wiley-VCH: Weinheim, Germany, 2000. |
| 1. | Szejtli, J. Chem. Rev. 1998, 98, 1743–1754. doi:10.1021/cr970022c |
| 27. | Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183 |
| 22. | Menuel, S.; Saitzek, S.; Monflier, E.; Hapiot, F. Beilstein J. Org. Chem. 2020, 16, 2598–2606. doi:10.3762/bjoc.16.211 |
| 23. | Menuel, S.; Doumert, B.; Saitzek, S.; Ponchel, A.; Delevoye, L.; Monflier, E.; Hapiot, F. J. Org. Chem. 2015, 80, 6259–6266. doi:10.1021/acs.joc.5b00697 |
| 14. | Jicsinszky, L.; Iványi, R. Carbohydr. Polym. 2001, 45, 139–145. doi:10.1016/s0144-8617(00)00319-2 |
| 24. | Cravotto, G.; Caporaso, M.; Jicsinszky, L.; Martina, K. Beilstein J. Org. Chem. 2016, 12, 278–294. doi:10.3762/bjoc.12.30 |
| 25. | Jicsinszky, L.; Rossi, F.; Solarino, R.; Cravotto, G. Molecules 2023, 28, 467. doi:10.3390/molecules28020467 |
| 48. | Reddy, L. R.; Reddy, M. A.; Bhanumathi, N.; Rao, K. R. Indian J. Chem., Sect. B 2002, 41B, 645–646. |
| 8. | Petter, R. C.; Salek, J. S.; Sikorski, C. T.; Kumaravel, G.; Lin, F.-T. J. Am. Chem. Soc. 1990, 112, 3860–3868. doi:10.1021/ja00166a021 |
| 21. | Martina, K.; Trotta, F.; Robaldo, B.; Belliardi, N.; Jicsinszky, L.; Cravotto, G. Tetrahedron Lett. 2007, 48, 9185–9189. doi:10.1016/j.tetlet.2007.10.104 |
| 47. | Jones, R. V.; Godorhazy, L.; Varga, N.; Szalay, D.; Urge, L.; Darvas, F. J. Comb. Chem. 2006, 8, 110–116. doi:10.1021/cc050107o |
© 2023 Orosz et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.