Abstract
Pyrrolo[2,1-b][1,3]benzothiazoles are an important class of fused sulfur and nitrogen-containing heterocycles intensively studied in medicinal chemistry and pharmacology. In the present paper, a new synthetic approach to pyrrolobenzothiazoles is developed based on 1,4-thiazine ring contraction in 3-aroylpyrrolo[2,1-c][1,4]benzothiazine-1,2,4-triones under the action of nucleophiles. The proposed approach works well with alkanols, benzylamine, and arylamines. The scope and limitations of the developed approach are studied. The synthesized pyrrolobenzothiazole derivatives represent an interest to pharmaceutics, since their close analogs show CENP-E inhibitory activity, interesting for the targeted cancer therapy development.

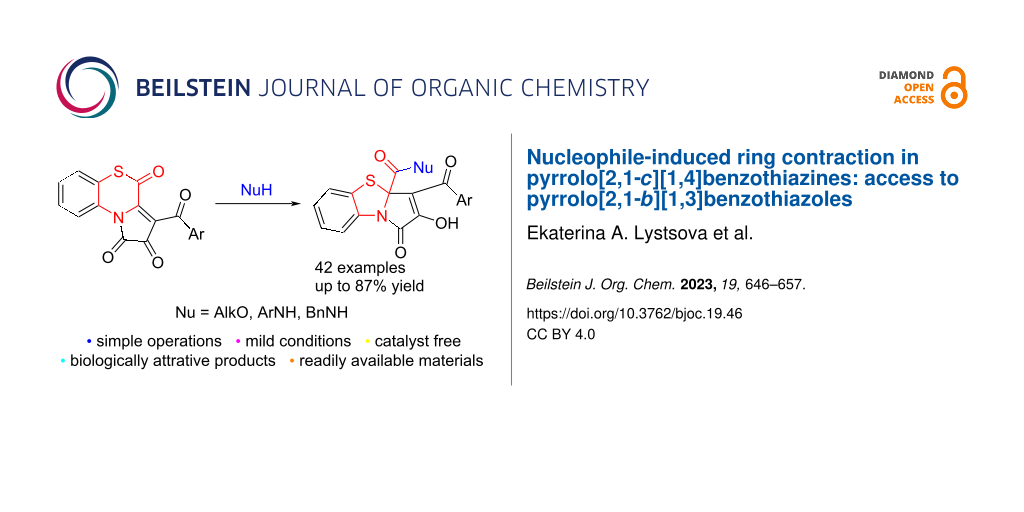
Graphical Abstract
Introduction
Pyrrolo[2,1-b][1,3]benzothiazole (PBTA) is an angularly fused sulfur and nitrogen-containing heterocyclic scaffold. Its derivatives are popular in medicinal chemistry and pharmacology as potential biologically active compounds. In particular, PBTAs were found to be promising inhibitors of centromere-associated protein E (CENP-E) (Figure 1), which is demanded for the development of targeted cancer therapy [1]. Furthermore, candidate anticonvulsant agents had been developed based on PBTA derivatives (Figure 1) [2]. In addition, series of PBTAs (Figure 1) were found to exhibit antibacterial, antifungal, antioxidant, and cytotoxic activities [3,4].
![[1860-5397-19-46-1]](/bjoc/content/figures/1860-5397-19-46-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Several strategies have been developed for the synthesis of PBTA derivatives [2-32] to meet the needs of medicinal chemistry and pharmacology for PBTAs containing diverse substituents. In general, these synthetic strategies can be divided into four groups.
The first group of approaches to the PBTA scaffold is an annulation of benzothiazoles with a pyrrole moiety (Scheme 1). It includes intramolecular cyclizations of benzothiazoles bearing a 3'-chloro substituent at C2 position (Scheme 1, entries 1 and 2) [5-7], intramolecular catalytic carbene cascade reactions of propargyl 1,3-benzothiazol-2-yl(diazo)acetates (Scheme 1, entry 3) [12], dearomative [3 + 2] cycloaddition reactions of benzothiazoles with cyclopropanes (Scheme 1, entry 4) [13-15], multicomponent reactions (MCRs) of benzothiazoles, isocyanides and 2-methylidenemalonates (Scheme 1, entry 5) [16], 1,3-dipolar cycloadditions of N-alkylbenzothiazolium salts (Scheme 1, entry 6) [17-22], MCRs of 2-methylbenzothiazole, acetylenedicarboxylates and active methylene compounds (Scheme 1, entry 7) [23-25], MCRs of (1,3-benzothiazol-2-yl)acetonitrile, aldehydes and acylcyanides (Scheme 1, enry 8) [3,26] and reactions of 3-acyl-2,3-dihydro-1,3-benzothiazole-2-carbonitriles with acetylenedicarboxylate (Scheme 1, entry 9) [4].
![[1860-5397-19-46-i1]](/bjoc/content/inline/1860-5397-19-46-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Approaches to PBTAs via annulation of benzothiazoles.
Scheme 1: Approaches to PBTAs via annulation of benzothiazoles.
The second group of approaches to the PBTA scaffold is an annulation of o-aminothiophenol with a pyrrolothiazole moiety (Scheme 2). It includes catalytic cascade reactions of o-aminothiophenol with donor–acceptor cyclopropanes (Scheme 2, entry 10) [27], condensations of o-aminothiophenol with 4-oxo acids or their derivatives (Scheme 2, entry 11) [2,28-31] and cascade reactions of o-aminothiophenol, furfural and anhydrides of 2,3-unsaturated carboxylic acids (Scheme 2, entry 12) [32].
![[1860-5397-19-46-i2]](/bjoc/content/inline/1860-5397-19-46-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Approaches to PBTAs via annulation of o-aminothiophenols.
Scheme 2: Approaches to PBTAs via annulation of o-aminothiophenols.
The third group of approaches to the PBTA scaffold includes only one example, the intramolecular radical substitution reaction in 1-(2-bromophenyl)-5-(butylsulfanyl)pyrrolidin-2-one (Scheme 3, entry 13) [8].
![[1860-5397-19-46-i3]](/bjoc/content/inline/1860-5397-19-46-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Approach to PBTAs via radical substitution reaction in 1-(2-bromophenyl)-5-(butylsulfanyl)pyrrolidin-2-one.
Scheme 3: Approach to PBTAs via radical substitution reaction in 1-(2-bromophenyl)-5-(butylsulfanyl)pyrrolidi...
The fourth group of approaches to the PBTA scaffold is the intramolecular cyclization of 1-(2-thiophenyl)pyrroles (Scheme 4). It includes intramolecular cationic π-cyclizations in 3-hydroxy-2-(2-sulfanylphenyl)-2,3-dihydro-1H-isoindol-1-ones (Scheme 4, entry 14) [9] and intramolecular cyclizations of 1-(2-(methylsulfinyl)phenyl)-1H-pyrroles under «interrupted Pummerer rearrangement» conditions (Scheme 4, entry 15) [10,11].
![[1860-5397-19-46-i4]](/bjoc/content/inline/1860-5397-19-46-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Approach to PBTAs via intramolecular cyclizations of 1-(2-thiophenyl)pyrroles.
Scheme 4: Approach to PBTAs via intramolecular cyclizations of 1-(2-thiophenyl)pyrroles.
This work reports a new approach to PBTA derivatives via nucleophile-induced ring contraction in pyrrolo[2,1-c][1,4]benzothiazines 1 (Scheme 5, entry 16), which can generally be attributed as a new entry to the fourth group of approaches to the PBTA scaffold (Scheme 4).
![[1860-5397-19-46-i5]](/bjoc/content/inline/1860-5397-19-46-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A new approach to PBTAs via nucleophile-induced ring contraction in pyrrolo[2,1-c][1,4]benzothiazines.
Scheme 5: A new approach to PBTAs via nucleophile-induced ring contraction in pyrrolo[2,1-c][1,4]benzothiazin...
Results and Discussion
It is known that [e]-fused 1H-pyrrole-2,3-diones (FPDs) (Figure 2) are versatile synthetic platforms enabling the synthesis of numerous heterocyclic species [33-36]. They are polyelectrophilic compounds, bearing five electrophilic centers, whose reactivity dramatically depends on the nature of the heteroatom X in FPDs I, II, 1 [33,34].
![[1860-5397-19-46-2]](/bjoc/content/figures/1860-5397-19-46-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
It should be noted that the reactions of 5-aza- and 5-oxa-FPDs I and II with nucleophiles are studied rather well [33,34], and their reactivity did not give us any insights for the development of new approaches to PBTAs. However, recently, we have reported a new class of FPDs, aroylpyrrolobenzothiazinetriones (APBTTs) 1 (Figure 2) [37,38], whose structural features allowed us to assume a possibility of the development of a new approach to PBTAs via a nucleophile-induced ring contraction in the 1,4-benzothiazine moiety of compounds 1 (Scheme 5). Firstly, FPDs 1 bear a 1,4-benzothiazine moiety that is known to be prone to undergo a ring contraction reaction to afford the corresponding 1,3-benzothiazole derivatives under the action of nucleophiles [39-42], oxidizing agents [43-48] or ultraviolet irradiation [49]. Secondly, the presence of a highly reactive thioester group C4=O [50] in FPDs 1 made us to expect the position C4 (Figure 2) to be the most reactive electrophilic center in these molecules, which would also contribute to the development of a new synthetic approach to PBTAs.
We started our research by conducting a test reaction of APBTT 1a with anhydrous methanol 2a (Scheme 6). As a result, we obtained the expected PBTA 3aa in a good isolated yield (52%). The product 3aa was isolated by simple recrystallization of the evaporated reaction mixture.
![[1860-5397-19-46-i6]](/bjoc/content/inline/1860-5397-19-46-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of APBTT 1a with methanol (2a).
Scheme 6: Reaction of APBTT 1a with methanol (2a).
Apparently, the reaction proceeded according to the plausible pathway shown in Scheme 6. As we expected, the nucleophile 2a attacked on the position C4 of the substrate 1a, which resulted in the cleavage of the S5–C4 bond and the formation of a thiol intermediate A (1-(2-thiophenyl)pyrrole derivative generated in situ as a precursor analog for approaches from Scheme 4). Then, intermediate A underwent an intramolecular cyclization by the attack of the SH group on the C5 atom of the pyrrole-2,3-dione moiety to afford intermediate B that underwent a 1,3-prototropic shift to give product 3aa.
Next, the conditions (Table 1) of the model reaction of APBTT 1a with methanol (2a) were optimized. The best yield of PBTA 3aa was observed when methanol was used both as a solvent and a reagent and heated at 65 °C for 1 h (entry 7, Table 1). It is useful to note that, under these conditions, an increase in the heating time (up to 6 h) did not affect the HPLC-UV yield of compound 3aa. These conditions were taken as a standard for further reactions.
Table 1: Reaction of APBTT 1a with methanol (2a) in different solvents.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-46-i22.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Conditions | |
| Ab | Bc | ||
| Yield,d % | |||
| 1 | acetone | 13 | 17 |
| 2 | acetonitrile | 38 | 20 |
| 3 | butyl acetate | 16 | 29 |
| 4 | chloroform | 27 | 29 |
| 5 | 1,4-dioxane | 33 | 29 |
| 6 | hexane | 35e | 39f |
| 7 | methanol | 64 | 81 |
| 8 | toluene | 23 | 10 |
aReaction scale: a mixture of 1a (10 mg, 29.8 µmol), solvent (500 µL), and 2a (1.2 µL, 29.8 µmol) was stirred in an oven-dried closed microreaction V-vial. bConditions A: room temperature, 24 h. cConditions B: heating at the boiling point temperature of the solvent, 1 h. dHPLC–UV yields (biphenyl was used as an internal standard; each entry was carried out in triplicate, and the yields are given as mean values). eReaction was monitored for 14 days. fReaction time was 5 h.
Noteworthy, we had to derivatize product 3aa to detect it by HPLC–UV during the optimization studies. For these purposes, compound 3aa was converted to compound 4 by an earlier procedure developed by us (Scheme 7) [51]. Without this derivatization procedure, we could not accurately detect compound 3aa by HPLC–UV, since the chromatographic signals of untreated compound 3aa were broad and blurry.
![[1860-5397-19-46-i7]](/bjoc/content/inline/1860-5397-19-46-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, the reactant scope of the reaction was explored by involving to the reaction APBTTs 1a–h, bearing various aroyl substituents, and anhydrous alcohols 2a–c (Scheme 8) [52].
![[1860-5397-19-46-i8]](/bjoc/content/inline/1860-5397-19-46-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reaction of APBTTs 1a–h with alcohols 2a–c. Isolated yields are given; reaction scale: a mixture of 1 (0.45 mmol) and alcohol 2 (5 mL) was stirred in an oven-dried closed microreaction V-vial at the boiling point temperature of the used alcohol 2.
Scheme 8: Reaction of APBTTs 1a–h with alcohols 2a–c. Isolated yields are given; reaction scale: a mixture of ...
As a result, we found that the proposed procedure afforded target products 3 in poor to very good isolated yields (Scheme 8). We also observed that the nature of the aroyl substituents in substrates 1a–h did not significantly affect the yields of the corresponding products 3 and the general course of the reaction. However, the structure of the alcohols 2a–c had an effect on the studied reaction. Reactions with isopropyl alcohol 2b required longer reaction times (UPLC–UV–MS monitoring). This phenomenon could be due to the steric factors brought in by a bulky isopropyl substituent in alcohol 2b.
In addition, in all studied cases we observed that the reaction of APBTTs 1 with alcohols 2 always afforded labile side-products 5 (Scheme 9). Compounds 5 were formed when the nucleophile 2 attacked on the position C3a of the substrates 1. Such a direction of nucleophilic addition of alcohols to FPD species was observed earlier on the example of 5-oxa-FPDs II and was found to be reversible [33,53,54].
![[1860-5397-19-46-i9]](/bjoc/content/inline/1860-5397-19-46-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Side-reaction of APBTTs 1 with alcohols 2.
Scheme 9: Side-reaction of APBTTs 1 with alcohols 2.
We isolated products 5a,b,e to study their chemical behavior in solutions. We found that when compounds 5a,b,e were dissolved in anhydrous solvents (toluene, acetonitrile, DMSO-d6) at room temperature, or when these solutions were slightly heated, the compounds 5a,b,e dissociated to form APBTTs 1 (the solutions got violet color, characteristic of compounds 1) (Scheme 10). In the presence of water (including the atmospheric moisture), hydration products 6a,b,e were formed (Scheme 10). These observations are in a full accordance with the studies of similar products of 5-oxa-FPDs II [33,53,54].
![[1860-5397-19-46-i10]](/bjoc/content/inline/1860-5397-19-46-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Transformations of compounds 5 in solutions.
Scheme 10: Transformations of compounds 5 in solutions.
We assume compounds 5 to be products of the kinetic control of the reaction, and compounds 3, of the thermodynamic one. In addition, the formation of compounds 5 is reversible, and the formation of compounds 3 is irreversible. These assumptions were proved experimentally. In the study of the reaction of APBTT 1a with methanol (2a) by UPLC–UV–MS, we found that in 5 min at room temperature, the reaction mixture contained about 90% of product 5a, and in 1 h of heating the reaction mixture at 65 °C, it contained trace amounts of product 5a and 81 % of product 3aa.
Additionally, we have examined the scaling of the reaction of APBTT 1a with methanol (2a). We found that the proposed procedure could be readily scaled up to 1.5 mmol (0.5 g) of APBTT 1a. The isolated yield of compound 3aa was 50%. However, under such conditions, a longer reaction time was required (about 3 h, UPLC–UV–MS monitoring).
Then, to expand the scope of the developed approach to PBTAs, we examined several more groups of nucleophilic reagents.
For these purposes, we carried out a test reaction of APBTT 1a with benzylamine (Scheme 11). As we expected, this reaction proceeded similarly to the reaction of APBTTs 1 with alcohols 2, and we obtained the desired PBTA 7a in a good isolated yield (44%). The product 7a was isolated by simple crystallization from the reaction mixture.
![[1860-5397-19-46-i11]](/bjoc/content/inline/1860-5397-19-46-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Reaction of APBTT 1a with benzylamine.
Scheme 11: Reaction of APBTT 1a with benzylamine.
Next, the conditions (Table 2) of the model reaction of APBTT 1a and benzylamine were optimized. The best yield of PBTA 7a was observed when acetonitrile was used as the solvent and heated at 85 °C for 3 h (entry 2, Table 2). Since the product 7a isolation procedure proceeded more conveniently in toluene (the product could be isolated by simple filtration directly from the reaction mixture), and the yield of PBTA 7a was satisfactory, we chose these conditions (entry 7, Table 2) as a standard for further reactions.
Table 2: Reaction of APBTT 1a with benzylamine in different solvents.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-46-i23.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Conditions | |
| Ab | Bc | ||
| Yield,d % | |||
| 1 | acetone | 31 | 35 |
| 2 | acetonitrile | 69 | 87 |
| 3 | butyl acetate | 45 | 61 |
| 4 | chloroform | 61 | 68 |
| 5 | 1,4-dioxane | 32 | 53 |
| 6 | hexane | tracese | tracesf |
| 7 | toluene | 40 | 50 |
aReaction scale: a mixture of 1a (10 mg, 29.8 µmol), solvent (500 µL), and benzylamine (3.3 µL, 29.8 µmol) was stirred in an oven-dried closed microreaction V-vial. bConditions A: room temperature, 24 h. cConditions B: heating at the boiling point temperature of the solvent, 3 h. dHPLC–UV yields (biphenyl was used as an internal standard; each entry was carried out in triplicate, and the yields are given as mean values). eReaction was monitored for 10 days. fReaction time was 8 h.
As in the case of the above studied reaction with alcohols (Scheme 7), we had to derivatize product 7a (Scheme 12) by an earlier procedure developed by us [51] in order to investigate the reaction optimization by HPLC–UV.
![[1860-5397-19-46-i12]](/bjoc/content/inline/1860-5397-19-46-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Then, the reactant scope of the reaction was explored by involving to the reaction APBTTs 1a–h, bearing various aroyl substituents, and benzylamine (Scheme 13) [55].
![[1860-5397-19-46-i13]](/bjoc/content/inline/1860-5397-19-46-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Reaction of APBTTs 1a–h and benzylamine. Isolated yields are given; reaction scale: a mixture of 1 (0.45 mmol), benzylamine (0.49 mmol, 54 µL) and anhydrous toluene (3 mL) was stirred in an oven-dried closed microreaction V-vial.
Scheme 13: Reaction of APBTTs 1a–h and benzylamine. Isolated yields are given; reaction scale: a mixture of 1 ...
As a result, we found that the proposed procedure afforded target products 7 in poor to good isolated yields (Scheme 13). We also observed that the nature of the aroyl substituents in substrates 1a–h did not significantly affect the yields of the corresponding products 7 and the general course of the reaction.
We also examined the influence of an excess of benzylamine on the yields of PBTAs 7. In the reaction of APBTT 1a with benzylamine in a ratio of 1:2 (reaction conditions were the same as in Scheme 13), the isolated yield of the product 7a was lower (35%) than in the case of the reaction in a ratio of 1:1.1 (Scheme 13). However, when conducting the reaction in a ratio of 1:10 at room temperature during 24 h, we observed the formation of N1,N2-dibenzyloxalamide (9) as a major product (NMR yield of about 90%) (Scheme 14).
![[1860-5397-19-46-i14]](/bjoc/content/inline/1860-5397-19-46-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Reaction of APBTT 1a with an excess of benzylamine.
Scheme 14: Reaction of APBTT 1a with an excess of benzylamine.
Our attempts to employ other alkylamines (diethylamine, morpholine, and cyclohexylamine) to the proposed approach to PBTAs were not successful. In the reaction of APBTT 1a with diethylamine (1a/diethylamine ratio of 1:1; stirring in toluene at 90 °С for 2 h; at 113 °С for 2 h; at room temperature for 24 h) and cyclohexylamine (1a/cyclohexylamine ratio of 1:1 or 1:5; stirring in toluene at room temperature for 24 h), a mixture of unidentified substances was formed. Interestingly, in the reaction of APBTT 1a with morpholine, we succeeded to isolate the product 10a (Scheme 15) [56]. Product 10a was formed in a result of a nucleophilic attack of morpholine on the C1 position of APBTT 1a.
![[1860-5397-19-46-i15]](/bjoc/content/inline/1860-5397-19-46-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Reaction of APBTT 1a with morpholine.
Scheme 15: Reaction of APBTT 1a with morpholine.
Such a change in the reaction selectivity could be explained by the influence of a higher nucleophilicity of the examined alkylamines in comparison with benzylamine.
Then, we tried to involve less nucleophilic amines to the proposed approach.
For these, we examined a reaction of APBTT 1a with aniline (11a, Scheme 16). This reaction proceeded similarly to reactions of APBTTs 1 with alcohols 2 and benzylamine, and we obtained the desired PBTA 12aa in a moderate isolated yield (40%). The product 12aa was isolated by simple crystallization from the reaction mixture.
![[1860-5397-19-46-i16]](/bjoc/content/inline/1860-5397-19-46-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Reaction of APBTT 1a with aniline (11a).
Scheme 16: Reaction of APBTT 1a with aniline (11a).
Next, the conditions (Table 3) of the model reaction of APBTT 1a and aniline (11a) were optimized. The best yield of PBTA 12aa was observed when butyl acetate was used as the solvent and heated at 130 °C for 3 h (entry 3, Table 3). Heating the reaction mixture in toluene (entry 7, Table 3) showed a good yield too. Since the product 12aa isolation procedure proceeded more conveniently in toluene, we chose these conditions (entry 7, Table 3) as a standard for further reactions.
Table 3: Reaction of APBTT 1a with aniline (11a) in different solvents.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-46-i24.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Conditions | |
| Ab | Bc | ||
| Yield,d % | |||
| 1 | acetone | 40 | 0 |
| 2 | acetonitrile | 37 | 37 |
| 3 | butyl acetate | 34 | 83 |
| 4 | chloroform | 44 | 35 |
| 5 | 1,4-dioxane | 44 | 47 |
| 6 | hexane | 40e | 32f |
| 7 | toluene | 36 | 73 |
aReaction scale: a mixture of 1a (10 mg, 29.8 µmol), solvent (500 µL), and 11a (2.7 µL, 29.8 µmol) was stirred in an oven-dried closed microreaction V-vial. bConditions A: room temperature, 24 h. cConditions B: heating at the boiling point temperature of the solvent, 7 h. dHPLC–UV yields. eReaction was monitored for 14 days. fReaction time was 6 h.
As in the cases of the above studied reactions (Scheme 7, Scheme 12), we had to derivatize the product 12aa (Scheme 17) by an earlier procedure developed by us [51] in order to investigate the reaction optimization by HPLC-UV.
![[1860-5397-19-46-i17]](/bjoc/content/inline/1860-5397-19-46-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
After that, the reactant scope of the reaction was explored by involving to the reaction APBTTs 1a–h, bearing various aroyl substituents, and arylamines 11a–d, bearing aryl substituents with various electronic effects (Scheme 18).
![[1860-5397-19-46-i18]](/bjoc/content/inline/1860-5397-19-46-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Reaction of APBTTs 1a–h and arylamines 11a–d. Isolated yields are given; reaction scale: a mixture of 1 (0.45 mmol), arylamine (0.45 mmol or 0.49 mmol), and anhydrous toluene (3–4 mL) was stirred in an oven-dried closed microreaction V-vial. aReaction time was 3 h for 11d and 8 h for 11a–c. bMes = 2,4,6-Me3C6H2.
Scheme 18: Reaction of APBTTs 1a–h and arylamines 11a–d. Isolated yields are given; reaction scale: a mixture ...
As a result, we have found that the proposed procedure afforded target products 12 in poor to good isolated yields (Scheme 18). We also observed that the nature of the aroyl substituents in substrates 1a–h and aryl substituents in amines 11a–c did not significantly affect the yields of the corresponding products 12 and the general course of the reaction. However, the reaction with o-aminoacetanilide 11d required shorter reaction times (visual monitoring of the reaction mixture color change and the precipitate formation) and afforded products 12 with higher isolated yields, which could probably be due to the solubility of the starting amine 11d and the corresponding products 12.
In addition, in the reaction of APBTT 1a with mesidine (11b), we succeeded to isolate a side-product 14ab (Scheme 19). Similar side-products 14 were observed in all reactions of APBTTs 1 with arylamines 11 (UPLC–UV–MS monitoring).
![[1860-5397-19-46-i19]](/bjoc/content/inline/1860-5397-19-46-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Side-reaction of APBTT 1a with arylamine 11b.
Scheme 19: Side-reaction of APBTT 1a with arylamine 11b.
Apparently, the reaction of APBTTs 1 with examined amines (benzylamine, alkylamines, arylamines 11) proceeded simultaneously in several directions: initial nucleophilic attack on positions C1, C2 or C4 of compounds 1. The ratio of yields of competitive reaction products depended on the nucleophilicity of the amine.
Then, we examined the proposed approach to PBTAs by involving bulky nucleophilic compounds 16a–d to the reaction with APBTT 1a. Unexpectedly, product 17a was formed instead of the anticipated PBTA C (Scheme 20). Compound 17a was formed in good isolated yields (60–70%) and was isolated by a simple crystallization from the reaction mixture.
![[1860-5397-19-46-i20]](/bjoc/content/inline/1860-5397-19-46-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Reaction of APBTT 1a with compounds 16a–d.
Scheme 20: Reaction of APBTT 1a with compounds 16a–d.
Moreover, we observed the formation of compounds 17 during acylation of enamines 15 with oxalyl chloride to prepare the starting APBTTs 1 (Scheme 21). We noticed that in this case, compounds 17 were formed when HCl was not effectively removed from the reaction mixtures. Bubbling of anhydrous argon through the reaction mixtures facilitated the removal of HCl, and reduced the formation of products 17 to trace amounts. Because of this, we assume that the formation of compounds 17 was caused in the result of addition of HCl to APBTTs 1 to form intermediates D [57], which underwent intramolecular nucleophilic substitution of the chloro substituent at C3a position with S5 [58] to give intermediates E. Then, intermediates E readily decarbonylated [59,60] to afford compounds 17 (Scheme 21). We suppose that in the reaction of APBTTs 1 with nucleophiles 16a–d, the formation of compounds 17 could proceed via a similar pathway, since Nu-groups of compounds 16a–d are good bulky leaving groups for nucleophilic substitution reactions. Nevertheless, the pathway of formation of compounds 17 is questionable and may become a subject of a new study.
![[1860-5397-19-46-i21]](/bjoc/content/inline/1860-5397-19-46-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Formation of compounds 17 as an undesired process during the synthesis of APBTTs 1.
Scheme 21: Formation of compounds 17 as an undesired process during the synthesis of APBTTs 1.
Conclusion
In conclusion, we have developed a new approach to pyrrolo[2,1-b][1,3]benzothiazoles 3, 7, and 12 via nucleophilic transformations of 3-aroylpyrrolo[2,1-c][1,4]benzothiazine-1,2,4-triones 1. The studied process presents a nucleophile-induced 1,4-benzothiazine ring contraction in compounds 1 through the cleavage of the S–C bond of the 1,4-benzothiazine moiety under the action of the nucleophile to form in situ a 1-(2-thiophenyl)pyrrole derivative that undergoes an intramolecular cyclization to give the target pyrrolobenzothiazoles 3, 7, and 12. The developed approach works well with alkanols 2, benzylamine, and arylamines 11, while alkylamines are unsuitable for it. Notable, the use of bulky nucleophiles (tert-butyl alcohol (16a), benzyl alcohol (16b), benzhydrol (16c), 2-aminobenzothiazole (16d), HCl) makes it possible to obtain pyrrolobenzothiazoles 17 from compounds 1, but their formation proceeds through a different pathway from the one to pyrrolobenzothiazoles 3, 7, and 12.
Supporting Information
| Supporting Information File 1: Further experimental details, copies of NMR spectra, X-ray crystallographic details, optimization by HPLC-UV details. | ||
| Format: PDF | Size: 4.8 MB | Download |
| Supporting Information File 2: Crystallographic information files (CIF) of compounds 3bb (CCDC 2241415), 4 (CCDC 2241420), 6b (CCDC 2241419), 6e (CCDC 2241423), 7a (CCDC 2241418), 10a (CCDC 2241424), 12bd (CCDC 2241422), 14ab (CCDC 2241416), 17a (CCDC 2241417, 2241421). | ||
| Format: ZIP | Size: 1.2 MB | Download |
References
-
Yamane, M.; Sawada, J.-i.; Ogo, N.; Ohba, M.; Ando, T.; Asai, A. Biochem. Biophys. Res. Commun. 2019, 519, 505–511. doi:10.1016/j.bbrc.2019.09.028
Return to citation in text: [1] -
Trapani, G.; Franco, M.; Latrofa, A.; Genchi, G.; Siro Brigiani, G.; Mazzoccoli, M.; Persichella, M.; Serra, M.; Biggio, G.; Liso, G. Eur. J. Med. Chem. 1994, 29, 197–204. doi:10.1016/0223-5234(94)90038-8
Return to citation in text: [1] [2] [3] -
Al-Mutairi, A. A.; Hafez, H. N.; El-Gazzar, A.-R. B. A.; Mohamed, M. Y. A. Molecules 2022, 27, 1246. doi:10.3390/molecules27041246
Return to citation in text: [1] [2] [3] -
Chaniyara, R.; Tala, S.; Chen, C.-W.; Lee, P.-C.; Kakadiya, R.; Dong, H.; Marvania, B.; Chen, C.-H.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Eur. J. Med. Chem. 2012, 53, 28–40. doi:10.1016/j.ejmech.2012.03.030
Return to citation in text: [1] [2] [3] -
Volovenko, Y. M.; Tverdokhlebov, A. V.; Volovnenko, T. A. Chem. Heterocycl. Compd. 2001, 37, 876–884. doi:10.1023/a:1012407826196
Return to citation in text: [1] [2] -
Mohamed, K. S.; Abdulaziz, N. M.; Fadda, A. A. J. Heterocycl. Chem. 2013, 50, 650–653. doi:10.1002/jhet.1685
Return to citation in text: [1] [2] -
Shokol, T. V.; Gorbulenko, N. V.; Khilya, V. P. Chem. Heterocycl. Compd. 2019, 55, 469–472. doi:10.1007/s10593-019-02482-w
Return to citation in text: [1] [2] -
Beckwith, A. L. J.; Boate, D. R. J. Org. Chem. 1988, 53, 4339–4348. doi:10.1021/jo00253a028
Return to citation in text: [1] [2] -
Hucher, N.; Decroix, B.; Daïch, A. J. Org. Chem. 2001, 66, 4695–4703. doi:10.1021/jo0156316
Return to citation in text: [1] [2] -
Bates, D. K.; Winters, R. T.; Picard, J. A. J. Org. Chem. 1992, 57, 3094–3097. doi:10.1021/jo00037a027
Return to citation in text: [1] [2] -
Boyd, S.; Davies, R. D. M.; Degorce, S. L.; Groombridge, S.; Scott, J. S.; Stokes, S. Tetrahedron Lett. 2016, 57, 152–154. doi:10.1016/j.tetlet.2015.11.083
Return to citation in text: [1] [2] -
Zhang, C.; Chang, S.; Qiu, L.; Xu, X. Chem. Commun. 2016, 52, 12470–12473. doi:10.1039/c6cc06864b
Return to citation in text: [1] [2] -
Wang, D.-C.; Xie, M.-S.; Guo, H.-M.; Qu, G.-R.; Zhang, M.-C.; You, S.-L. Angew. Chem., Int. Ed. 2016, 55, 14111–14115. doi:10.1002/anie.201607852
Return to citation in text: [1] [2] -
Preindl, J.; Chakrabarty, S.; Waser, J. Chem. Sci. 2017, 8, 7112–7118. doi:10.1039/c7sc03197a
Return to citation in text: [1] [2] -
Zhang, M.-C.; Wang, D.-C.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M.; You, S.-L. Chem 2019, 5, 156–167. doi:10.1016/j.chempr.2018.10.003
Return to citation in text: [1] [2] -
Xiong, Q.; Li, G.; Dong, S.; Liu, X.; Feng, X. Org. Lett. 2019, 21, 8771–8775. doi:10.1021/acs.orglett.9b03389
Return to citation in text: [1] [2] -
Kucukdisli, M.; Opatz, T. Eur. J. Org. Chem. 2012, 4555–4564. doi:10.1002/ejoc.201200424
Return to citation in text: [1] [2] -
Shen, G.-L.; Sun, J.; Yan, C.-G. Org. Biomol. Chem. 2015, 13, 10929–10938. doi:10.1039/c5ob01374g
Return to citation in text: [1] [2] -
Shen, G.; Sun, J.; Yan, C. Chin. J. Chem. 2016, 34, 412–418. doi:10.1002/cjoc.201500896
Return to citation in text: [1] [2] -
Jing, S.; Gong, J.; Chaoguo, Y.; Ying, H. Synthesis method of benzo[d]pyrrolo[2,1-b]thiazole. Chin. Patent CN106866706A, June 20, 2017.
Return to citation in text: [1] [2] -
Jiang, W.; Sun, J.; Yan, C.-G. RSC Adv. 2017, 7, 42387–42392. doi:10.1039/c7ra06548e
Return to citation in text: [1] [2] -
Zeng, Q.; Ren, D.; Fu, X.; Li, X. J. Chem. Res. 2018, 42, 260–263. doi:10.3184/174751918x15260567362969
Return to citation in text: [1] [2] -
Yavari, I.; Piltan, M.; Moradi, L. Tetrahedron 2009, 65, 2067–2071. doi:10.1016/j.tet.2009.01.001
Return to citation in text: [1] [2] -
Nassiri, M.; Milani, F. J.; Hassankhani, A. J. Heterocycl. Chem. 2015, 52, 1162–1166. doi:10.1002/jhet.2208
Return to citation in text: [1] [2] -
Milani, F. J.; Nassiri, M. J. Heterocycl. Chem. 2017, 54, 1840–1844. doi:10.1002/jhet.2774
Return to citation in text: [1] [2] -
Martinez-Ariza, G.; Mehari, B. T.; Pinho, L. A. G.; Foley, C.; Day, K.; Jewett, J. C.; Hulme, C. Org. Biomol. Chem. 2017, 15, 6076–6079. doi:10.1039/c7ob01239j
Return to citation in text: [1] [2] -
Chang, F.; Shen, B.; Wang, S.; Lin, L.; Feng, X. Chem. Commun. 2020, 56, 13429–13432. doi:10.1039/d0cc05667g
Return to citation in text: [1] [2] -
Khattab, S. N.; Hassan, S. Y.; El-Faham, A.; El Massry, A. M. M.; Amer, A. J. Heterocycl. Chem. 2007, 44, 617–626. doi:10.1002/jhet.5570440317
Return to citation in text: [1] [2] -
Cayley, A. N.; Gallagher, K. A.; Ménard-Moyon, C.; Schmidt, J. P.; Diorazio, L. J.; Taylor, R. J. K. Synthesis 2008, 3846–3856. doi:10.1055/s-0028-1083214
Return to citation in text: [1] [2] -
Sashidhara, K. V.; Singh, L. R.; Palnati, G. R.; Avula, S. R.; Kant, R. Synlett 2016, 27, 2384–2390. doi:10.1055/s-0035-1562614
Return to citation in text: [1] [2] -
Grinev, V. S.; Linkova, E. I.; Vasilchenko, D. S.; Egorova, A. Y. J. Struct. Chem. 2019, 60, 1688–1692. doi:10.1134/s0022476619100159
Return to citation in text: [1] [2] -
Zubkov, F. I.; Nikitina, E. V.; Galeev, T. R.; Zaytsev, V. P.; Khrustalev, V. N.; Novikov, R. A.; Orlova, D. N.; Varlamov, A. V. Tetrahedron 2014, 70, 1659–1690. doi:10.1016/j.tet.2014.01.008
Return to citation in text: [1] [2] -
Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889
Return to citation in text: [1] [2] [3] [4] [5] -
Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204
Return to citation in text: [1] [2] [3] -
Lystsova, E. A.; Khramtsova, E. E.; Maslivets, A. N. Symmetry 2021, 13, 1509. doi:10.3390/sym13081509
Return to citation in text: [1] -
Topanov, P. A.; Maslivets, A. A.; Dmitriev, M. V.; Mashevskaya, I. V.; Shklyaev, Y. V.; Maslivets, A. N. Beilstein J. Org. Chem. 2022, 18, 1532–1538. doi:10.3762/bjoc.18.162
Return to citation in text: [1] -
Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Beilstein J. Org. Chem. 2020, 16, 2322–2331. doi:10.3762/bjoc.16.193
Return to citation in text: [1] -
Khramtsova, E. E.; Lystsova, E. A.; Dmitriev, M. V.; Maslivets, A. N.; Jasiński, R. ChemistrySelect 2021, 6, 6295–6301. doi:10.1002/slct.202101990
Return to citation in text: [1] -
Kollenz, G.; Seidler, P. Monatsh. Chem. 1984, 115, 623–628. doi:10.1007/bf00799171
Return to citation in text: [1] -
Sako, M.; Niwa, T.; Hirota, K.; Maki, Y. Chem. Pharm. Bull. 1986, 34, 664–668. doi:10.1248/cpb.34.664
Return to citation in text: [1] -
Georgiev, V. S.; Kropp, P. L.; Carlson, R. P.; Van Inwegen, R. G.; Mack, R. A. Eur. J. Med. Chem. 1989, 24, 639–641. doi:10.1016/0223-5234(89)90036-6
Return to citation in text: [1] -
Puebla, P.; Honores, Z.; Medarde, M.; Morán, L.; Caballero, E.; San Feliciano, A. Tetrahedron 1999, 55, 7915–7922. doi:10.1016/s0040-4020(99)00401-9
Return to citation in text: [1] -
Charrier, J.-D.; Landreau, C.; Deniaud, D.; Reliquet, F.; Reliquet, A.; Meslin, J. C. Tetrahedron 2001, 57, 4195–4202. doi:10.1016/s0040-4020(01)00317-9
Return to citation in text: [1] -
Chill, L.; Rudi, A.; Benayahu, Y.; Kashman, Y. Tetrahedron Lett. 2004, 45, 7925–7928. doi:10.1016/j.tetlet.2004.08.137
Return to citation in text: [1] -
Stalling, T.; Saak, W.; Martens, J. Eur. J. Org. Chem. 2013, 6291–6297. doi:10.1002/ejoc.201300768
Return to citation in text: [1] -
Blunt, C. E.; Nawrat, C. C.; LeBozec, L.; Liutkus, M.; Liu, Y.; Lewis, W.; Moody, C. J. Synlett 2015, 27, 37–40. doi:10.1055/s-0035-1560722
Return to citation in text: [1] -
Liao, S.-R.; Tang, Y.; Xu, L.; Zhou, X.-F.; Wang, J.-F.; Yang, B.; Liu, Y.-H. Tetrahedron 2017, 73, 98–107. doi:10.1016/j.tet.2016.11.062
Return to citation in text: [1] -
Nguyen, T. B.; Retailleau, P. Adv. Synth. Catal. 2020, 362, 5380–5384. doi:10.1002/adsc.202000964
Return to citation in text: [1] -
Costantini, C.; Testa, G.; Crescenzi, O.; d'Ischia, M. Tetrahedron Lett. 1994, 35, 3365–3366. doi:10.1016/s0040-4039(00)76909-7
Return to citation in text: [1] -
Shi, Y.; Liu, X.; Cao, H.; Bie, F.; Han, Y.; Yan, P.; Szostak, R.; Szostak, M.; Liu, C. Org. Biomol. Chem. 2021, 19, 2991–2996. doi:10.1039/d1ob00187f
Return to citation in text: [1] -
Khramtsova, E. E.; Lystsova, E. A.; Khokhlova, E. V.; Dmitriev, M. V.; Maslivets, A. N. Molecules 2021, 26, 7179. doi:10.3390/molecules26237179
Return to citation in text: [1] [2] [3] -
Maslivets, A. N.; Khramtsova, E. E.; Lystsova, E. A. Method for producing methyl 3-aroyl-2-hydroxy-1-oxobenzo[d]pyrrolo[2,1-b]thiazole-3a(1H)-carboxylates exhibiting antioxidant activity. Russ. Patent RU2785751 C1, Dec 12, 2022.
Return to citation in text: [1] -
Maslivets, A. N.; Mashevskaya, I. V.; Krasnykh, O. P.; Shurov, S. N.; Andreichikov, Y. S. Zh. Org. Khim. 1992, 28, 2545–2553.
Return to citation in text: [1] [2] -
Gumerova, D. F.; Mashevskaya, I. V.; Maslivets, A. N.; Kozlov, A. P. Russ. J. Org. Chem. 2003, 39, 995–997. doi:10.1023/b:rujo.0000003192.96463.ca
Return to citation in text: [1] [2] -
Maslivets, A. N.; Khramtsova, E. E.; Lystsova, E. A. Method for the preparation of 3-aryl-N-benzyl-2-hydroxy-1-oxobenzo[d]pyrrolo[2,1-b]thiazole-3a(1H)-carboxamides. Russ. Patent RU2764906 C1, Jan 24, 2022.
Return to citation in text: [1] -
Maslivets, A. N.; Khramtsova, E. E.; Lystsova, E. A. Method for the preparation of 4-aryl-1-morpholino-3-(2-oxo-2H-benzo[b][1,4]thiazine-3(4H)-ylidene)-butane-1,2,4-triones exhibiting fluorescent properties. Russ. Patent RU2782022 C1, Oct 21, 2022.
Return to citation in text: [1] -
Sabitov, A. A.; Khramtsova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2022, 58, 287–294. doi:10.1134/s1070428022030058
Return to citation in text: [1] -
Amosova, S. V.; Filippov, A. A.; Makhaeva, N. A.; Albanov, A. I.; Potapov, V. A. Beilstein J. Org. Chem. 2020, 16, 515–523. doi:10.3762/bjoc.16.47
Return to citation in text: [1] -
Schaumann, E.; Behrens, U. Angew. Chem., Int. Ed. Engl. 1977, 16, 722–723. doi:10.1002/anie.197707222
Return to citation in text: [1] -
Schaumann, E.; Lange, B.; Reinholdt, K. J. Chem. Soc., Chem. Commun. 1983, 797–798. doi:10.1039/c39830000797
Return to citation in text: [1]
| 52. | Maslivets, A. N.; Khramtsova, E. E.; Lystsova, E. A. Method for producing methyl 3-aroyl-2-hydroxy-1-oxobenzo[d]pyrrolo[2,1-b]thiazole-3a(1H)-carboxylates exhibiting antioxidant activity. Russ. Patent RU2785751 C1, Dec 12, 2022. |
| 33. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 53. | Maslivets, A. N.; Mashevskaya, I. V.; Krasnykh, O. P.; Shurov, S. N.; Andreichikov, Y. S. Zh. Org. Khim. 1992, 28, 2545–2553. |
| 54. | Gumerova, D. F.; Mashevskaya, I. V.; Maslivets, A. N.; Kozlov, A. P. Russ. J. Org. Chem. 2003, 39, 995–997. doi:10.1023/b:rujo.0000003192.96463.ca |
| 33. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 53. | Maslivets, A. N.; Mashevskaya, I. V.; Krasnykh, O. P.; Shurov, S. N.; Andreichikov, Y. S. Zh. Org. Khim. 1992, 28, 2545–2553. |
| 54. | Gumerova, D. F.; Mashevskaya, I. V.; Maslivets, A. N.; Kozlov, A. P. Russ. J. Org. Chem. 2003, 39, 995–997. doi:10.1023/b:rujo.0000003192.96463.ca |
| 1. | Yamane, M.; Sawada, J.-i.; Ogo, N.; Ohba, M.; Ando, T.; Asai, A. Biochem. Biophys. Res. Commun. 2019, 519, 505–511. doi:10.1016/j.bbrc.2019.09.028 |
| 5. | Volovenko, Y. M.; Tverdokhlebov, A. V.; Volovnenko, T. A. Chem. Heterocycl. Compd. 2001, 37, 876–884. doi:10.1023/a:1012407826196 |
| 6. | Mohamed, K. S.; Abdulaziz, N. M.; Fadda, A. A. J. Heterocycl. Chem. 2013, 50, 650–653. doi:10.1002/jhet.1685 |
| 7. | Shokol, T. V.; Gorbulenko, N. V.; Khilya, V. P. Chem. Heterocycl. Compd. 2019, 55, 469–472. doi:10.1007/s10593-019-02482-w |
| 32. | Zubkov, F. I.; Nikitina, E. V.; Galeev, T. R.; Zaytsev, V. P.; Khrustalev, V. N.; Novikov, R. A.; Orlova, D. N.; Varlamov, A. V. Tetrahedron 2014, 70, 1659–1690. doi:10.1016/j.tet.2014.01.008 |
| 59. | Schaumann, E.; Behrens, U. Angew. Chem., Int. Ed. Engl. 1977, 16, 722–723. doi:10.1002/anie.197707222 |
| 60. | Schaumann, E.; Lange, B.; Reinholdt, K. J. Chem. Soc., Chem. Commun. 1983, 797–798. doi:10.1039/c39830000797 |
| 2. | Trapani, G.; Franco, M.; Latrofa, A.; Genchi, G.; Siro Brigiani, G.; Mazzoccoli, M.; Persichella, M.; Serra, M.; Biggio, G.; Liso, G. Eur. J. Med. Chem. 1994, 29, 197–204. doi:10.1016/0223-5234(94)90038-8 |
| 3. | Al-Mutairi, A. A.; Hafez, H. N.; El-Gazzar, A.-R. B. A.; Mohamed, M. Y. A. Molecules 2022, 27, 1246. doi:10.3390/molecules27041246 |
| 4. | Chaniyara, R.; Tala, S.; Chen, C.-W.; Lee, P.-C.; Kakadiya, R.; Dong, H.; Marvania, B.; Chen, C.-H.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Eur. J. Med. Chem. 2012, 53, 28–40. doi:10.1016/j.ejmech.2012.03.030 |
| 5. | Volovenko, Y. M.; Tverdokhlebov, A. V.; Volovnenko, T. A. Chem. Heterocycl. Compd. 2001, 37, 876–884. doi:10.1023/a:1012407826196 |
| 6. | Mohamed, K. S.; Abdulaziz, N. M.; Fadda, A. A. J. Heterocycl. Chem. 2013, 50, 650–653. doi:10.1002/jhet.1685 |
| 7. | Shokol, T. V.; Gorbulenko, N. V.; Khilya, V. P. Chem. Heterocycl. Compd. 2019, 55, 469–472. doi:10.1007/s10593-019-02482-w |
| 8. | Beckwith, A. L. J.; Boate, D. R. J. Org. Chem. 1988, 53, 4339–4348. doi:10.1021/jo00253a028 |
| 9. | Hucher, N.; Decroix, B.; Daïch, A. J. Org. Chem. 2001, 66, 4695–4703. doi:10.1021/jo0156316 |
| 10. | Bates, D. K.; Winters, R. T.; Picard, J. A. J. Org. Chem. 1992, 57, 3094–3097. doi:10.1021/jo00037a027 |
| 11. | Boyd, S.; Davies, R. D. M.; Degorce, S. L.; Groombridge, S.; Scott, J. S.; Stokes, S. Tetrahedron Lett. 2016, 57, 152–154. doi:10.1016/j.tetlet.2015.11.083 |
| 12. | Zhang, C.; Chang, S.; Qiu, L.; Xu, X. Chem. Commun. 2016, 52, 12470–12473. doi:10.1039/c6cc06864b |
| 13. | Wang, D.-C.; Xie, M.-S.; Guo, H.-M.; Qu, G.-R.; Zhang, M.-C.; You, S.-L. Angew. Chem., Int. Ed. 2016, 55, 14111–14115. doi:10.1002/anie.201607852 |
| 14. | Preindl, J.; Chakrabarty, S.; Waser, J. Chem. Sci. 2017, 8, 7112–7118. doi:10.1039/c7sc03197a |
| 15. | Zhang, M.-C.; Wang, D.-C.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M.; You, S.-L. Chem 2019, 5, 156–167. doi:10.1016/j.chempr.2018.10.003 |
| 16. | Xiong, Q.; Li, G.; Dong, S.; Liu, X.; Feng, X. Org. Lett. 2019, 21, 8771–8775. doi:10.1021/acs.orglett.9b03389 |
| 17. | Kucukdisli, M.; Opatz, T. Eur. J. Org. Chem. 2012, 4555–4564. doi:10.1002/ejoc.201200424 |
| 18. | Shen, G.-L.; Sun, J.; Yan, C.-G. Org. Biomol. Chem. 2015, 13, 10929–10938. doi:10.1039/c5ob01374g |
| 19. | Shen, G.; Sun, J.; Yan, C. Chin. J. Chem. 2016, 34, 412–418. doi:10.1002/cjoc.201500896 |
| 20. | Jing, S.; Gong, J.; Chaoguo, Y.; Ying, H. Synthesis method of benzo[d]pyrrolo[2,1-b]thiazole. Chin. Patent CN106866706A, June 20, 2017. |
| 21. | Jiang, W.; Sun, J.; Yan, C.-G. RSC Adv. 2017, 7, 42387–42392. doi:10.1039/c7ra06548e |
| 22. | Zeng, Q.; Ren, D.; Fu, X.; Li, X. J. Chem. Res. 2018, 42, 260–263. doi:10.3184/174751918x15260567362969 |
| 23. | Yavari, I.; Piltan, M.; Moradi, L. Tetrahedron 2009, 65, 2067–2071. doi:10.1016/j.tet.2009.01.001 |
| 24. | Nassiri, M.; Milani, F. J.; Hassankhani, A. J. Heterocycl. Chem. 2015, 52, 1162–1166. doi:10.1002/jhet.2208 |
| 25. | Milani, F. J.; Nassiri, M. J. Heterocycl. Chem. 2017, 54, 1840–1844. doi:10.1002/jhet.2774 |
| 26. | Martinez-Ariza, G.; Mehari, B. T.; Pinho, L. A. G.; Foley, C.; Day, K.; Jewett, J. C.; Hulme, C. Org. Biomol. Chem. 2017, 15, 6076–6079. doi:10.1039/c7ob01239j |
| 27. | Chang, F.; Shen, B.; Wang, S.; Lin, L.; Feng, X. Chem. Commun. 2020, 56, 13429–13432. doi:10.1039/d0cc05667g |
| 28. | Khattab, S. N.; Hassan, S. Y.; El-Faham, A.; El Massry, A. M. M.; Amer, A. J. Heterocycl. Chem. 2007, 44, 617–626. doi:10.1002/jhet.5570440317 |
| 29. | Cayley, A. N.; Gallagher, K. A.; Ménard-Moyon, C.; Schmidt, J. P.; Diorazio, L. J.; Taylor, R. J. K. Synthesis 2008, 3846–3856. doi:10.1055/s-0028-1083214 |
| 30. | Sashidhara, K. V.; Singh, L. R.; Palnati, G. R.; Avula, S. R.; Kant, R. Synlett 2016, 27, 2384–2390. doi:10.1055/s-0035-1562614 |
| 31. | Grinev, V. S.; Linkova, E. I.; Vasilchenko, D. S.; Egorova, A. Y. J. Struct. Chem. 2019, 60, 1688–1692. doi:10.1134/s0022476619100159 |
| 32. | Zubkov, F. I.; Nikitina, E. V.; Galeev, T. R.; Zaytsev, V. P.; Khrustalev, V. N.; Novikov, R. A.; Orlova, D. N.; Varlamov, A. V. Tetrahedron 2014, 70, 1659–1690. doi:10.1016/j.tet.2014.01.008 |
| 8. | Beckwith, A. L. J.; Boate, D. R. J. Org. Chem. 1988, 53, 4339–4348. doi:10.1021/jo00253a028 |
| 3. | Al-Mutairi, A. A.; Hafez, H. N.; El-Gazzar, A.-R. B. A.; Mohamed, M. Y. A. Molecules 2022, 27, 1246. doi:10.3390/molecules27041246 |
| 4. | Chaniyara, R.; Tala, S.; Chen, C.-W.; Lee, P.-C.; Kakadiya, R.; Dong, H.; Marvania, B.; Chen, C.-H.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Eur. J. Med. Chem. 2012, 53, 28–40. doi:10.1016/j.ejmech.2012.03.030 |
| 27. | Chang, F.; Shen, B.; Wang, S.; Lin, L.; Feng, X. Chem. Commun. 2020, 56, 13429–13432. doi:10.1039/d0cc05667g |
| 57. | Sabitov, A. A.; Khramtsova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2022, 58, 287–294. doi:10.1134/s1070428022030058 |
| 2. | Trapani, G.; Franco, M.; Latrofa, A.; Genchi, G.; Siro Brigiani, G.; Mazzoccoli, M.; Persichella, M.; Serra, M.; Biggio, G.; Liso, G. Eur. J. Med. Chem. 1994, 29, 197–204. doi:10.1016/0223-5234(94)90038-8 |
| 2. | Trapani, G.; Franco, M.; Latrofa, A.; Genchi, G.; Siro Brigiani, G.; Mazzoccoli, M.; Persichella, M.; Serra, M.; Biggio, G.; Liso, G. Eur. J. Med. Chem. 1994, 29, 197–204. doi:10.1016/0223-5234(94)90038-8 |
| 28. | Khattab, S. N.; Hassan, S. Y.; El-Faham, A.; El Massry, A. M. M.; Amer, A. J. Heterocycl. Chem. 2007, 44, 617–626. doi:10.1002/jhet.5570440317 |
| 29. | Cayley, A. N.; Gallagher, K. A.; Ménard-Moyon, C.; Schmidt, J. P.; Diorazio, L. J.; Taylor, R. J. K. Synthesis 2008, 3846–3856. doi:10.1055/s-0028-1083214 |
| 30. | Sashidhara, K. V.; Singh, L. R.; Palnati, G. R.; Avula, S. R.; Kant, R. Synlett 2016, 27, 2384–2390. doi:10.1055/s-0035-1562614 |
| 31. | Grinev, V. S.; Linkova, E. I.; Vasilchenko, D. S.; Egorova, A. Y. J. Struct. Chem. 2019, 60, 1688–1692. doi:10.1134/s0022476619100159 |
| 58. | Amosova, S. V.; Filippov, A. A.; Makhaeva, N. A.; Albanov, A. I.; Potapov, V. A. Beilstein J. Org. Chem. 2020, 16, 515–523. doi:10.3762/bjoc.16.47 |
| 17. | Kucukdisli, M.; Opatz, T. Eur. J. Org. Chem. 2012, 4555–4564. doi:10.1002/ejoc.201200424 |
| 18. | Shen, G.-L.; Sun, J.; Yan, C.-G. Org. Biomol. Chem. 2015, 13, 10929–10938. doi:10.1039/c5ob01374g |
| 19. | Shen, G.; Sun, J.; Yan, C. Chin. J. Chem. 2016, 34, 412–418. doi:10.1002/cjoc.201500896 |
| 20. | Jing, S.; Gong, J.; Chaoguo, Y.; Ying, H. Synthesis method of benzo[d]pyrrolo[2,1-b]thiazole. Chin. Patent CN106866706A, June 20, 2017. |
| 21. | Jiang, W.; Sun, J.; Yan, C.-G. RSC Adv. 2017, 7, 42387–42392. doi:10.1039/c7ra06548e |
| 22. | Zeng, Q.; Ren, D.; Fu, X.; Li, X. J. Chem. Res. 2018, 42, 260–263. doi:10.3184/174751918x15260567362969 |
| 3. | Al-Mutairi, A. A.; Hafez, H. N.; El-Gazzar, A.-R. B. A.; Mohamed, M. Y. A. Molecules 2022, 27, 1246. doi:10.3390/molecules27041246 |
| 26. | Martinez-Ariza, G.; Mehari, B. T.; Pinho, L. A. G.; Foley, C.; Day, K.; Jewett, J. C.; Hulme, C. Org. Biomol. Chem. 2017, 15, 6076–6079. doi:10.1039/c7ob01239j |
| 56. | Maslivets, A. N.; Khramtsova, E. E.; Lystsova, E. A. Method for the preparation of 4-aryl-1-morpholino-3-(2-oxo-2H-benzo[b][1,4]thiazine-3(4H)-ylidene)-butane-1,2,4-triones exhibiting fluorescent properties. Russ. Patent RU2782022 C1, Oct 21, 2022. |
| 16. | Xiong, Q.; Li, G.; Dong, S.; Liu, X.; Feng, X. Org. Lett. 2019, 21, 8771–8775. doi:10.1021/acs.orglett.9b03389 |
| 4. | Chaniyara, R.; Tala, S.; Chen, C.-W.; Lee, P.-C.; Kakadiya, R.; Dong, H.; Marvania, B.; Chen, C.-H.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Eur. J. Med. Chem. 2012, 53, 28–40. doi:10.1016/j.ejmech.2012.03.030 |
| 51. | Khramtsova, E. E.; Lystsova, E. A.; Khokhlova, E. V.; Dmitriev, M. V.; Maslivets, A. N. Molecules 2021, 26, 7179. doi:10.3390/molecules26237179 |
| 13. | Wang, D.-C.; Xie, M.-S.; Guo, H.-M.; Qu, G.-R.; Zhang, M.-C.; You, S.-L. Angew. Chem., Int. Ed. 2016, 55, 14111–14115. doi:10.1002/anie.201607852 |
| 14. | Preindl, J.; Chakrabarty, S.; Waser, J. Chem. Sci. 2017, 8, 7112–7118. doi:10.1039/c7sc03197a |
| 15. | Zhang, M.-C.; Wang, D.-C.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M.; You, S.-L. Chem 2019, 5, 156–167. doi:10.1016/j.chempr.2018.10.003 |
| 51. | Khramtsova, E. E.; Lystsova, E. A.; Khokhlova, E. V.; Dmitriev, M. V.; Maslivets, A. N. Molecules 2021, 26, 7179. doi:10.3390/molecules26237179 |
| 12. | Zhang, C.; Chang, S.; Qiu, L.; Xu, X. Chem. Commun. 2016, 52, 12470–12473. doi:10.1039/c6cc06864b |
| 23. | Yavari, I.; Piltan, M.; Moradi, L. Tetrahedron 2009, 65, 2067–2071. doi:10.1016/j.tet.2009.01.001 |
| 24. | Nassiri, M.; Milani, F. J.; Hassankhani, A. J. Heterocycl. Chem. 2015, 52, 1162–1166. doi:10.1002/jhet.2208 |
| 25. | Milani, F. J.; Nassiri, M. J. Heterocycl. Chem. 2017, 54, 1840–1844. doi:10.1002/jhet.2774 |
| 55. | Maslivets, A. N.; Khramtsova, E. E.; Lystsova, E. A. Method for the preparation of 3-aryl-N-benzyl-2-hydroxy-1-oxobenzo[d]pyrrolo[2,1-b]thiazole-3a(1H)-carboxamides. Russ. Patent RU2764906 C1, Jan 24, 2022. |
| 33. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 34. | Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204 |
| 35. | Lystsova, E. A.; Khramtsova, E. E.; Maslivets, A. N. Symmetry 2021, 13, 1509. doi:10.3390/sym13081509 |
| 36. | Topanov, P. A.; Maslivets, A. A.; Dmitriev, M. V.; Mashevskaya, I. V.; Shklyaev, Y. V.; Maslivets, A. N. Beilstein J. Org. Chem. 2022, 18, 1532–1538. doi:10.3762/bjoc.18.162 |
| 9. | Hucher, N.; Decroix, B.; Daïch, A. J. Org. Chem. 2001, 66, 4695–4703. doi:10.1021/jo0156316 |
| 10. | Bates, D. K.; Winters, R. T.; Picard, J. A. J. Org. Chem. 1992, 57, 3094–3097. doi:10.1021/jo00037a027 |
| 11. | Boyd, S.; Davies, R. D. M.; Degorce, S. L.; Groombridge, S.; Scott, J. S.; Stokes, S. Tetrahedron Lett. 2016, 57, 152–154. doi:10.1016/j.tetlet.2015.11.083 |
| 50. | Shi, Y.; Liu, X.; Cao, H.; Bie, F.; Han, Y.; Yan, P.; Szostak, R.; Szostak, M.; Liu, C. Org. Biomol. Chem. 2021, 19, 2991–2996. doi:10.1039/d1ob00187f |
| 51. | Khramtsova, E. E.; Lystsova, E. A.; Khokhlova, E. V.; Dmitriev, M. V.; Maslivets, A. N. Molecules 2021, 26, 7179. doi:10.3390/molecules26237179 |
| 43. | Charrier, J.-D.; Landreau, C.; Deniaud, D.; Reliquet, F.; Reliquet, A.; Meslin, J. C. Tetrahedron 2001, 57, 4195–4202. doi:10.1016/s0040-4020(01)00317-9 |
| 44. | Chill, L.; Rudi, A.; Benayahu, Y.; Kashman, Y. Tetrahedron Lett. 2004, 45, 7925–7928. doi:10.1016/j.tetlet.2004.08.137 |
| 45. | Stalling, T.; Saak, W.; Martens, J. Eur. J. Org. Chem. 2013, 6291–6297. doi:10.1002/ejoc.201300768 |
| 46. | Blunt, C. E.; Nawrat, C. C.; LeBozec, L.; Liutkus, M.; Liu, Y.; Lewis, W.; Moody, C. J. Synlett 2015, 27, 37–40. doi:10.1055/s-0035-1560722 |
| 47. | Liao, S.-R.; Tang, Y.; Xu, L.; Zhou, X.-F.; Wang, J.-F.; Yang, B.; Liu, Y.-H. Tetrahedron 2017, 73, 98–107. doi:10.1016/j.tet.2016.11.062 |
| 48. | Nguyen, T. B.; Retailleau, P. Adv. Synth. Catal. 2020, 362, 5380–5384. doi:10.1002/adsc.202000964 |
| 49. | Costantini, C.; Testa, G.; Crescenzi, O.; d'Ischia, M. Tetrahedron Lett. 1994, 35, 3365–3366. doi:10.1016/s0040-4039(00)76909-7 |
| 37. | Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Beilstein J. Org. Chem. 2020, 16, 2322–2331. doi:10.3762/bjoc.16.193 |
| 38. | Khramtsova, E. E.; Lystsova, E. A.; Dmitriev, M. V.; Maslivets, A. N.; Jasiński, R. ChemistrySelect 2021, 6, 6295–6301. doi:10.1002/slct.202101990 |
| 39. | Kollenz, G.; Seidler, P. Monatsh. Chem. 1984, 115, 623–628. doi:10.1007/bf00799171 |
| 40. | Sako, M.; Niwa, T.; Hirota, K.; Maki, Y. Chem. Pharm. Bull. 1986, 34, 664–668. doi:10.1248/cpb.34.664 |
| 41. | Georgiev, V. S.; Kropp, P. L.; Carlson, R. P.; Van Inwegen, R. G.; Mack, R. A. Eur. J. Med. Chem. 1989, 24, 639–641. doi:10.1016/0223-5234(89)90036-6 |
| 42. | Puebla, P.; Honores, Z.; Medarde, M.; Morán, L.; Caballero, E.; San Feliciano, A. Tetrahedron 1999, 55, 7915–7922. doi:10.1016/s0040-4020(99)00401-9 |
| 33. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 34. | Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204 |
| 33. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 34. | Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204 |
© 2023 Lystsova et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.