Abstract
A set of novel 1,4-diaryl-1,3-butadiynes terminated by two 7-(arylethynyl)-1,8-bis(dimethylamino)naphthalene fragments was prepared via the Glaser–Hay oxidative dimerization of 2-ethynyl-7-(arylethynyl)-1,8-bis(dimethylamino)naphthalenes. The oligomers synthesized in this way are cross-conjugated systems, in which two conjugation pathways are possible: π-conjugation of 1,8-bis(dimethylamino)naphthalene (DMAN) fragments through a butadiyne linker and a donor–acceptor aryl–C≡C–DMAN conjugation path. The conjugation path can be “switched” simply by protonation of DMAN fragments. X-ray diffraction, UV–vis spectroscopy and cyclic voltammetry are applied to analyze the extent of π-conjugation and the efficiency of particular donor–acceptor conjugation path in these new compounds. X-ray structures and absorption spectra of doubly protonated tetrafluoroborate salts of the oligomers are also discussed.

Graphical Abstract
Introduction
π-Conjugated oligomers and polymers attracted considerable attention from the very start as a promising class of semiconductors, chemosensors, and various electronic devices [1,2]. Although silicon and inorganic materials still play a major role in the development of modern electronics, the prospects for using organic electronic materials as an alternative are becoming increasingly clear. One of the advantages of those materials is a possibility to fine-tune useful properties by simply varying of the π-conjugated backbone and side-chain substituents [1-5].
π-Conjugated oligomers consisting of alternating C≡C bonds and aromatic nuclei, commonly, have a rigid, rod-like structure and exhibit high charge carriers’ mobility [6]. 1,4-Diaryl-1,3-butadiynes is a particular class of such compounds. Both theoretical and experimental studies revealed that the side groups of 1,4-diaryl-1,3-butadiynes have a significant impact on their useful characteristics [7-14]. For example, single-molecule conductivity, nonlinear optical properties, and the ability to serve as photosensitizers of singlet oxygen production have been identified in porphyrin-based butadiynes [7-9], 1,3-butadiyne-linked oligoporphycenes [10], and 1,3-butadiyne-linked amines [13]. A wide variety of applications was proposed for graphdiynes (2D allotropes of graphene), including electrocatalysts and energy devices, which exploit the carbon-rich nature, porous framework, and expanded π-electron system of these compounds [11]. And this is not a complete list.
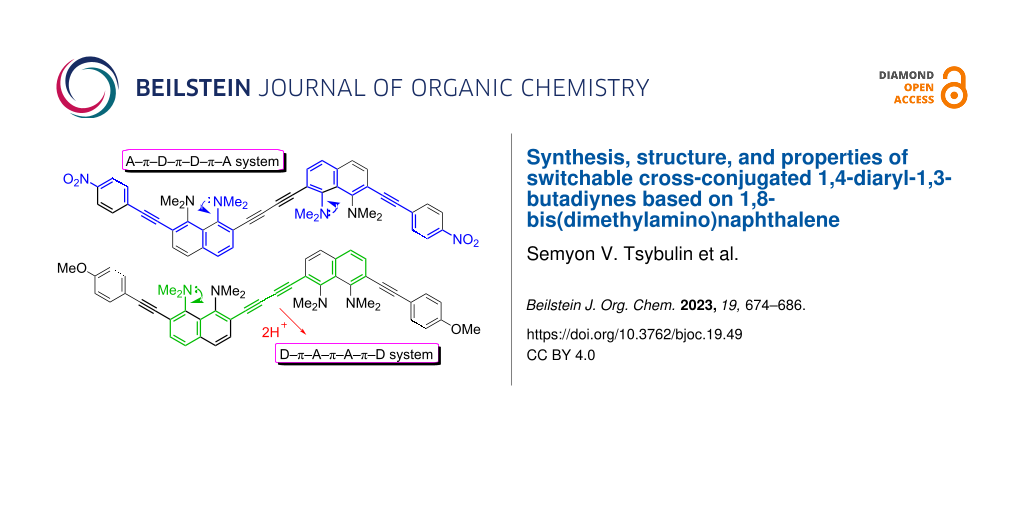
Recently, we reported on the synthesis of 1,4-diaryl-1,3-butadiynes 1–4 based on the “proton sponge” [1,8-bis(dimethylamino)naphthalene, DMAN] (Figure 1) [15]. In the present work we describe the synthesis of a new family of proton sponge-based butadiynes 5 bearing arylethynyl substituents of different electronic nature. Oligomers 5 having electron-withdrawing groups on the aryl termini are interesting as push–pull A–π–D–π–D–π–A systems, whereas the counterpart with an electron-donating methoxy group can be converted into a D–π–A–π–A–π–D system by protonation of the proton sponge fragments (Figure 2). Moreover, oligomers 5 are cross-conjugated π-systems. “A cross-conjugated compound may be defined as a compound possessing three unsaturated groups, two of which although conjugated to a third unsaturated center are not conjugated to each other” [16]. It is easy to see that there are two π-conjugation paths in molecules 5: a donor–acceptor conjugation path (Figure 2, highlighted in blue) and the π-conjugation of naphthalene rings through a butadiyne linker (highlighted in green). In comparison to linearly conjugated materials, oligomeric and polymeric compounds with a fully cross-conjugated carbon backbone are relatively unexplored [17-20]. Molecules of this type serve not only as objects of fundamental research into the phenomena of cross-conjugation, electron transfer, and quantum interference [17-20], but are also considered as promising molecular switches and transistors [21-25], NLO materials [26-29], and suitable starting compounds for syntheses involving multiple Diels–Alder additions [30]. All these facts motivated us to undertake the current study. X-ray crystallography, UV–vis spectroscopy and cyclic voltammetry were applied to analyze the extent of π-electron conjugation and the efficiency of the particular donor–acceptor conjugation path in chromophores 5.
![[1860-5397-19-49-1]](/bjoc/content/figures/1860-5397-19-49-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Proton sponge-based 1,4-diaryl-1,3-butadiynes synthesized previously and in this study.
Figure 1: Proton sponge-based 1,4-diaryl-1,3-butadiynes synthesized previously and in this study.
![[1860-5397-19-49-2]](/bjoc/content/figures/1860-5397-19-49-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Target oligomers as push–pull and cross-conjugated π-systems.
Figure 2: Target oligomers as push–pull and cross-conjugated π-systems.
Results and Discussion
Synthesis
The target oligomers 5 can be synthesized by a Glaser oxidative dimerization of monomers 6 (Scheme 1). The obvious route for the synthesis of the latter is the sequential alkynylation of 2,7-diiodonaphthalene 8.
![[1860-5397-19-49-i1]](/bjoc/content/inline/1860-5397-19-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic strategy for target oligomers 5.
Scheme 1: Synthetic strategy for target oligomers 5.
In accordance with this strategy, diiodide 8 was cross-coupled with copper(I) arylacetylides (Castro–Stephens reaction, method A) and arylacetylenes (Sonogashira reaction, method B). In all cases, even when using a small excess of 8, in addition to the desired monoalkynyl derivative 7, a double alkynylation product 9 was formed (Table 1). The Sonogashira coupling was somewhat more efficient, yielding compounds 7a–e in 42–62% yields, but also gave higher amounts of products 9a–e (10–30%). Thus, the Pd- and phosphine-free Castro–Stephens coupling was a good enough alternative to synthesize alkynes 7. The structure of the double alkynylation product 9e was confirmed by X-ray diffraction data (see Supporting Information, File 1, Figure S60).
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-49-i6.svg?max-width=637&scale=1.0)
The further alkynylation of compounds 7a–e was carried out using trimethylsilylacetylene and the Pd(PPh3)2Cl2/CuI/Et3N/DMSO catalytic system giving rise to dialkynyl derivatives 10a–e in high yields (Scheme 2). Column chromatography of trimethylsilyl derivatives 10a–e on Al2O3 resulted in their quantitative desilylation with the formation of the target monomers 6a–e, thus eliminating the need to remove the trimethylsilyl protection. Pure samples of compounds 10a–e could be obtained by extraction of the reaction mixture with hexane followed by recrystallization of the crude product from ethanol.
![[1860-5397-19-49-i2]](/bjoc/content/inline/1860-5397-19-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 7-(arylethynyl)-2-ethynyl-DMAN 6.
Scheme 2: Synthesis of 7-(arylethynyl)-2-ethynyl-DMAN 6.
Next, the oxidative dimerization of terminal alkynes 6a–e was carried out in an aerobic medium in the CuI/TMEDA/iPr2NH system at room temperature, which proved to be effective in the synthesis of butadiynes 1–4 [15] (Scheme 3). The desired diarylbutadiynes 5a–e were obtained in good yields regardless of the substituent R in the benzene ring. Treatment of the latter with fluoroboric acid in dichloromethane gave double salts 11a–e.
![[1860-5397-19-49-i3]](/bjoc/content/inline/1860-5397-19-49-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1,4-diaryl-1,3-butadiynes 5 and their salts 11.
Scheme 3: Synthesis of 1,4-diaryl-1,3-butadiynes 5 and their salts 11.
X-ray structures
Slow evaporation of solutions of butadiynes 5 in the CHCl3/EtOAc system made it possible to grow single crystals of samples 5b, 5d, and 5e suitable for X-ray diffraction studies (Figure 3 and Figure 4). Crystals of compound 5c were grown up using CHCl3/EtOH solvent, and it was unexpectedly found that keeping this compound in the above system for a month leads to its partial heterocyclization to benzo[g]indole 12 (Scheme 4 and Figure S59 in Supporting Information File 1). The structure of compound 12 was unambiguously established by X-ray diffraction analysis (see Supporting Information File 1, Figure S61). We assumed that this transformation is facilitated by hydrogen chloride, which is formed during the oxidation of chloroform with atmospheric oxygen. We also succeeded in growing crystals of the salt 11c in the MeCN/EtOH system (Figure 5). Unfortunately, good crystals of other salts have not been obtained.
![[1860-5397-19-49-3]](/bjoc/content/figures/1860-5397-19-49-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structures of compounds 5b (top), 5d (middle), and 5e (bottom).
Figure 3: Molecular structures of compounds 5b (top), 5d (middle), and 5e (bottom).
![[1860-5397-19-49-4]](/bjoc/content/figures/1860-5397-19-49-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Views on the molecular backbone of compounds 5b (top), 5d (middle), and 5e (bottom) along the naphthalene rings plane (hydrogen atoms omitted).
Figure 4: Views on the molecular backbone of compounds 5b (top), 5d (middle), and 5e (bottom) along the napht...
![[1860-5397-19-49-i4]](/bjoc/content/inline/1860-5397-19-49-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Transformation of butadiyne 5c into benzo[g]indole 12.
Scheme 4: Transformation of butadiyne 5c into benzo[g]indole 12.
![[1860-5397-19-49-5]](/bjoc/content/figures/1860-5397-19-49-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Molecular structure of compound 11c: frontal (top; BF4− omitted) and side views (bottom; hydrogen atoms omitted).
Figure 5: Molecular structure of compound 11c: frontal (top; BF4− omitted) and side views (bottom; hydrogen a...
From Figure 3 and Figure 4 it is easy to see that all molecules 5b, 5d, and 5e are rather distorted, including naphthalene cores, butadiyne and acetylene linkers. The main structural parameters of diynes 5 that characterize the degree of this distortion are presented in Table 2, where ϕ1 is the angle between the planes of the benzene ring and the neighboring naphthalene system, ϕ2 is the angle between the averaged planes of the naphthalene rings, ∠Cx‒Cy–Cz is the bond angle of the carbon–carbon bonds in the butadiyne linker, Θ is the C2(2′)–C3(3′)–C6(6′)–C7(7′) torsion, N···N is the internitrogen distance in the DMAN fragments, Σ∠N is the sum of the C–N–C angles of the NMe2 groups, and φ is the N1(1′)–C1(1′)–C8(8′)–N8(8′) torsion. For salt 11c, two additional parameters characterizing the hydrogen N–H···N bond are given, e.g., the N–H bond lengths and the angle between them (∠N–H···N).
Table 2: Some structural parameters of oligomers 5 and salt 11c (X-ray data).
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-49-i7.svg?max-width=637&scale=1.0)
|
||||||||
| Parameter | 5b (R = OMe) | 5d (R = CN) | 5e (R = NO2) | 11ca | ||||
| A | B | A | B | A = B | A = B | |||
| ϕ2 torsion, ° | 17.4 | 34.4 | 0 | 0 | 0 | |||
| ϕ1 torsion, ° | 1.4 | 12.7 | 28.2 | 29.0 | 17.9 | 32.4 | 12.8 | |
|
∠C2‒Cα‒Сβ, °
∠Cα‒Cβ‒Сγ, ° ∠Cβ‒Cγ‒Сδ, ° ∠Cγ‒Cδ‒С2′, ° |
176.9
172.9 176.0 170.2 |
177.5
177.7 177.9 176.7 |
172.2
177.6 |
173.5
178.2 |
176.1
177.7 |
|||
|
∠C7(7')‒Ca(a')‒Сb(b'), °
∠Ca(a')‒Cb(b')‒Сc(c'), ° |
173.2
178.3 |
173.3
179.1 |
174.1
178.5 |
174.5
176.9 |
174.5
172.2 |
168.5
174.2 |
175.0
176.2 |
|
| C2(2′)–C3(3′)–C6(6′)–C7(7′) torsion Θ, ° | 19.6 | 15.3 | 12.8 | 9.5 | 18.6 | 0.8 | 0.0 | |
| N···N distance, Å | 2.859 | 2.808 | 2.779 | 2.772 | 2.848 | 2.553 | 2.570 | |
| sum of the C–N–C angles Σ∠N, ° | N1(1′) | 357.3 | 357.6 | 354.4 | 354.9 | 357.6 | 340.0 | 340.5 |
| N8(8′) | 359.3 | 357.0 | 357.3 | 353.5 | 358.6 | 341.0 | 340.9 | |
| N1(1′)–C1(1′)–C8(8′)–N8(8′) torsion φ, ° | 29.0 | 21.7 | 20.9 | 8.4 | 31.6 | 2.7 | 2.5 | |
| N–H···N bond lengths, Å | – | – | – | – | – |
1.03
1.54 |
1.01
1.59 |
|
| ∠N–H···N, ° | – | – | – | – | – | 166 | 162 | |
aStructural parameters of two independent molecules are given.
In all cases naphthalene fragments linked by a 1,3-butadiyne axis take a trans position relative to each other. Despite formally symmetrical structure of diynes 5, naphthalene rings A and B of molecules 5b and 5d differ in their structural parameters. At the same time, the monomer fragments of the nitro derivative 5e are identical. In the case of 5e, the naphthalene rings lie in parallel planes, while in crystals of 5b and 5d the angle between the average planes of naphthalene nuclei A and B reaches 17° and 34°, respectively. Molecule 5d demonstrates the largest rotation angles of the aryl termini with respect to the naphthalene rings (ϕ1 = 28–29°). The dimethylamino groups of 5 are strongly flattened (the Σ∠N value varies from 353.5° to 359.3°), which is characteristic of ortho-substituted proton sponges [31]. The observed N···N distances are slightly larger than those typical for ortho-disubstituted DMAN derivatives [31].
Molecule 5b is the most distorted, as evidenced by the significant twisting of naphthalene rings (ΘA = 19.6° and ΘB = 15.3°), the largest internitrogen distance (2.859 and 2.808 Å for rings A and B, respectively), the largest deviation of the dimethylamino groups from the naphthalene ring plane (φA = 29.0°, φB = 21.7°), as well as deviation of the bond angles of the butadiyne linker from the standard value of 180° by 3–10°. Deviations of bond angles in acetylene bridges are ≈1–7°. The methoxy derivative 5b has the most complex crystal packing with a large number of different nonvalent interactions (see Supporting Information File 1, Figures S62 and S63).
The molecule of cyano derivative 5d is characterized by the least distortion of the DMAN fragments in the series (twisting Θ = 9.45 and 12.83°, torsions φA = 20.9° and φB = 8.4°, bond angle deviations in both butadiyne and acetylene linkers do not exceed 6°). In the crystal packing of 5d (see Supporting Information File 1, Figures S64 and S65), the DMAN fragments do not participate in nonvalent interactions and do not form short contacts. The recurring motif in the crystals is the coordination of the benzene meta proton by the nitrogen atom of the C≡N group.
The structural parameters of both monomer fragments of nitro derivative 5e are identical. This fact, together with the parallelism of the naphthalene ring planes, indicates the existence of an inversion center in the molecule. Molecule 5e is characterized by the largest N1(1′)–C1(1′)–C8(8′)–N8(8′) torsion angle φ (31.6°) in the series. Other structural parameters are close to those of the molecule 5b. In the crystal packing (Figure S66 in Supporting Information File 1), molecules 5e tend to approach π-donor DMAN and π-acceptor p-nitrophenyl fragments, and the shortest distance between the two molecules is 2.810 Å (Figure S67 in Supporting Information File 1).
The alternation of the C–C bond lengths in the aryl rings of molecules 5d and 5e may indirectly indicate the conjugation of the π-donor fragment with the π-acceptor p-nitrophenyl or p-cyanophenyl fragments. The qr parameter, calculated according to equation [32] (Figure 6) and characterizing the quinoid character of the aryl ring, was proposed for D–π–A systems. This parameter is a good indication for intramolecular charge transfer from the donor to the acceptor moiety in the ground state. In benzene, the qr value is equal to 0. In a fully quinoid ring, the qr was found to be equal to 0.10–0.12.
![[1860-5397-19-49-6]](/bjoc/content/figures/1860-5397-19-49-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Calculation of the qr parameter.
Figure 6: Calculation of the qr parameter.
Calculations based on the bond lengths in the aryl fragments determined by X-ray diffraction analysis gave the following average values of the qr parameter: 0.012 for 5d and 0.014 for 5e. For comparison, the same parameter calculated for N,N-dimethyl-4-nitroaniline is 0.038 (X-ray data from reference [33]). Therefore, the π-charge transfer from the donor DMAN to the acceptor aryl ring of 5d and 5e is extremely modest in the ground state. It should be also noted that the CNaph–N bonds of 5e are the shortest in the series (1.379–1.380 Å), which may also indirectly indicate a more pronounced conjugation of the dimethylamino groups with the nitro group.
There are two types of independent non-equivalent dications, marked in blue and green, and two types of BF4− anions, marked in red and yellow, in the crystal structure of salt 11c (Figure S68 in Supporting Information File 1). Monomer fragments in both are identical (Table 2, Figure 5). The trifluoromethyl group of one independent molecule is disordered with an occupancy of fluorine atoms of 0.54/0.46, which makes the molecule asymmetric. The second independent molecule has an inversion center. Compared to the free bases 5 discussed above, the protonated form 11с demonstrates almost complete planarization of the naphthalene backbone due to the disappearance of steric and electrostatic stress between the NMe2 groups. The nitrogen atoms in the DMAN fragments strongly approach during protonation and practically do not deviate from the average naphthalene ring plane. The dimethylamino groups naturally become more pyramidal and the CNaph–N bonds lengthen. All these changes are typical for protonated DMAN derivatives [31]. Interestingly, the NH protons are localized on the 1-NMe2 and 1′-NMe2 groups adjacent to the butadiyne linker and do not move away from each other at the maximum distance closer to the aryl substituents. Noteworthy is the bending of the acetylene linker (in one of the independent molecules, the bond angle is only 168.5°) and the greater linearity of the butadiyne fragment, which may indicate a more pronounced conjugation between two DMAN fragments than between DMAN and p-trifluoromethylphenyl rings. By the way, the qr parameters calculated for two independent molecules of 11c were 0.008 and 0.009, which is slightly less than in the case of compounds 5. As for the crystal packing of 11c, BF4− anions of two types (“red” and “yellow”) interact with cations in different ways. The “red” anion hangs over the cationic centers of both independent molecules, which are almost perpendicular to each other, while the “yellow” one participates mainly in the coordination with the hydrogen atoms of the NMe2 groups. Such a distribution of counterions apparently ensures mutually perpendicular packing of almost linear molecules (Figure S69, Supporting Information File 1).
UV–vis spectra and redox properties
As stated above, oligomers 5 are cross-conjugated π-systems. For cross-conjugated structures, the main question is about the preferential conjugation path. For oligomers 5, two different directions of electron density transfer are possible (Figure 7): between two DMAN fragments through the butadiyne linker (highlighted in green) and between the DMAN and aryl rings through the acetylene bridge (highlighted in blue). Obviously, the “butadiyne path” includes a longer conjugation chain. Noncovalent interactions of molecules in crystals and packing effects do not allow one to strictly judge the charge transfer in the oligomers 5. Therefore, we analyzed their UV–vis spectra (Table 3, Figure 8). The functional groups R are located at the far ends of the oligomeric chain and, from the steric point of view, cannot have a significant effect on conformational transformations of 5. All differences in optical properties must be of an electronic nature. It is obvious that the same “butadiyne conjugation pathway” (marked in green) is realized in compound 1, while the conjugation chain in monomers 6 is identical to the “blue” one in oligomers 5 (Figure 7). Thus, the UV–vis spectra of compounds 1 and 6 were used for comparison (Table 3, Figure 8).
![[1860-5397-19-49-7]](/bjoc/content/figures/1860-5397-19-49-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Two π-conjugation ways in oligomers 5.
Figure 7: Two π-conjugation ways in oligomers 5.
Table 3: Summary of the UV–vis spectraa of monomers 6, oligomers 5 (in CHCl3), and salts 11 (in MeCN).
| R | Monomer 6 | Oligomer 5 | Salt 11 | |||||
| λmax, nm (lg ε) | λmax, nm (lg ε) | λonset, nm | Egopt, eVb | λmax, nm (lg ε) | λonset, nm | Egopt, eVb | ||
| H | a | 393 (3.98) | 432 (4.51) | 519 | 2.39 | 382 sh (4.28) | 397 | 3.12 |
| OMe | b | 405 sh (3.92) | 423 (4.54) | 518 | 2.39 | 387 sh (4.31) | 415 | 2.99 |
| CF3 | c | 400 (4.08) | 437 (4.64) | 524 | 2.37 | 382 sh (4.30) | 390 | 3.18 |
| CN | d | 441 sh (4.08) | 449 (4.54) | 547 | 2.27 | 383 sh (4.24) | 392 | 3.16 |
| NO2 | e | 456 sh (3.92) | 453 (4.58) | 594 | 2.09 | 385 sh (4.19) | 396 | 3.13 |
aAbsorption maxima measured in the corresponding solutions at c = 10−5 M. bThe optical gap estimated from the onset point of the absorption spectra: Egopt = 1240/λonset.
![[1860-5397-19-49-8]](/bjoc/content/figures/1860-5397-19-49-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: UV–vis spectra of oligomers 5 (blue line), monomers 6 (red line), and butadiyne 1 (green line).
Figure 8: UV–vis spectra of oligomers 5 (blue line), monomers 6 (red line), and butadiyne 1 (green line).
The long-wave absorption maximum of the yellow-colored butadiyne 1 is observed at 429 nm (lg ε = 4.33) [15]. Compounds 5a‒с (R = H, OMe, CF3) are yellow, 5d (R = CN) is orange, and 5e (R = NO2) is a crimson crystalline substance. As can be seen from Table 3, compound 5b with electron-releasing methoxy groups shows the smallest λmax value, while the absorption maximum of the nitro derivative 5e is the most red-shifted in the series. There is a noticeable difference in the absorption maxima of diynes 5a–c and the corresponding monomers 6a–c. Moreover, the absorption maxima of compounds 5a (R = H), 5b (R = OMe), and 5c (R = CF3) are rather close to diyne 1. Figure 8 clearly demonstrates that in these cases the profiles of the long-wavelength maximum almost overlap with those of butadiyne 1. Apparently, in molecules 5a–c, the “butadiyne conjugation pathway” is realized, involving a larger number of multiple bonds. The absorption maxima of compounds 5a and 5c are slightly red-shifted (Δλ 3–8 nm), while in the case of compound 5b a hypsochromic shift of λmax is observed. On passing to oligomers 5d and 5e bearing strong electron-withdrawing CN and NO2 substituents in the benzene rings the picture changes. The π-deficient nature of the terminal aryl rings obviously facilitates their conjugation with π-excessive DMAN fragments. The λmax values and the general shape of the spectra of compounds 5d and 5e are closer to those of the corresponding monomers 6d and 6e, but not of butadiyne 1. The absorption maximum of derivative 5d is red-shifted by 8 nm relative to monomer 6d.
It should be noted that oligomers 5d and 5e as well as the corresponding monomers 6d and 6e are typical rod-like D–π–A systems. In such molecules, a photon absorption induces a shift from a D–π–A ground state to a D+–π–A− excited state. It is obvious that the actual electron transfer depends on all three components of the push–pull molecule. However, the study on the through-space charge transfer (CT) in the rod-like donor–acceptor molecules showed that adding a stronger electron-donating group does not systematically induce an enhancement of the CT if a strong electron-accepting moiety is used, the latter tending to extract the electron from the conjugated chains rather from the donor moiety [34]. We therefore compared the UV spectra of the oligomers 5d and 5e and monomers 6d and 6e with those of model p,p'-disubstituted diphenylacetylenes having donor NMe2 and acceptor NO2 or CN termini. The reported absorption maxima of 4-((4-(dimethylamino)phenyl)ethynyl)benzonitrile and N,N-dimethyl-4-((4-nitrophenyl)ethynyl)aniline in chloroform solution are 373 and 416 nm, respectively [35]. In the same time, λmax for 2-((4-nitrophenyl)ethynyl)-1,8-bis(dimethylamino)naphthalene is 474 nm [36]. The red shift observed in the spectrum of this compound as well as in the spectra of compounds 5d,e and 6d,e is likely a reflection of the elongation of the conjugation chain. This also supports the conjugation pathway between the nitro group and the more distant dimethylamino group of DMAN fragment marked in blue in Figure 7.
All synthesized oligomers 5 display no fluorescence in solution (chloroform and acetonitrile were tested). For comparison, N,N-dimethyl-4-((4-nitrophenyl)ethynyl)aniline demonstrates a weak fluorescence with an emission maximum at 550 nm (EtOH) [37]. The optical band gaps (Egopt), estimated from the onset point of the absorption spectra, ranged within 2.39 eV (for 5a and 5b) to 2.09 eV (for 5e). Thus, the HOMO–LUMO gap is significantly reduced by the introduction of the electron-withdrawing substituent, while the introduction of a donor substituent, e.g., a OMe group, does not change this value.
Previously, using the example of diphenylpolyynes containing a donor p-amino and an acceptor p’-nitro terminus, it was shown that upon protonation the band associated with the intramolecular charge-transfer transition emanating from the lone pair on the NH2 nitrogen and terminating in an empty π* orbital on the NO2 group, disappears [38]. The high-energy absorption for these compounds were largely unaffected by HCl protonation, the UV spectra of the protonated forms were very similar to those of unsubstituted diphenylpolyynes attributing the above bands to a π→π* transition. Сomplete protonation of DMAN-based diarylacetylenes led to a hypsochromic shift of their absorption maxima by 40–70 nm. The UV spectra of these salts are similar to those of the corresponding unsubstituted dinaphthylacetylenes [39].
In cases of oligomers 5 a comparable picture was observed. The protonated oligomers 11 show similar to each other UV–vis spectra and absorption maxima (Table 3, Figure 9). However, unlike salts 11a,c,d, methoxy derivative 11b demonstrates end absorption up to 415 nm and, thus, the lowest optical band gap in the series (2.99 eV). Evidently, protonation of 5b gives rise to a push–pull D–π–A–π–A–π–D system, in which the π-conjugation between the donating methoxyphenyl and the accepting protonated DMAN fragments becomes preferable (Figure 10). This is supported by a comparison of the absorption spectra of salt 11b and protonated forms of diyne 1 [15] and monomer 6b (salts 1·2HBF4 and 6b·HBF4), which demonstrates an obvious similarity of spectral curves of 11b and 6b·HBF4. Since the UV–vis spectra of salts 11 are similar (even and especially in cases of methoxy and nitro derivatives 11e and 11b), it can be assumed that in all salts 11 the electron transfer from the terminal aryl to the central naphthalene rings takes place. Naturally, the more deficient the aryl fragment, the less intense the long-wavelength maximum. Conjugation between the aryl substituent and the DMAN fragment in salts 11 is indirectly supported by the fact that the basicity of monomer 6b with the donor methoxy group (pKa = 8.2, measured in DMSO by the 1H NMR transprotonation approach [40]) is almost an order of magnitude higher than that of monomer 6e with the acceptor nitro group (pKa = 7.3).
![[1860-5397-19-49-9]](/bjoc/content/figures/1860-5397-19-49-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: UV–vis spectra of salts 11 (left), 1·2HBF4 and 6b·HBF4 (right) in acetonitrile.
Figure 9: UV–vis spectra of salts 11 (left), 1·2HBF4 and 6b·HBF4 (right) in acetonitrile.
![[1860-5397-19-49-10]](/bjoc/content/figures/1860-5397-19-49-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: π-Conjugation pathway in salts 11b and 6b·HBF4.
Figure 10: π-Conjugation pathway in salts 11b and 6b·HBF4.
The absorption maxima of 2,7-dialkynyl derivatives of DMAN 9 were observed at 402–465 nm, regularly shifting to the red region when passing from compound 9b bearing terminal methoxy groups to its analogs with electron-withdrawing substituents.
The redox properties of oligomers 5 were evaluated by cyclic voltammetry (CV) in dichloromethane solution containing 0.1 M n-Bu4NPF6 in the standard three-electrode electrochemical cell: glassy carbon working electrode, platinum auxiliary electrode, and reference electrode Ag/Ag+ 0.01 M AgNO3 (Figure 11, Table 4). Compounds 5a–d displayed two waves of irreversible oxidation in the potentials range of 0.0–1.1 V and one reduction wave (−1.5 to −1.6 V) with the little variation of the potentials induced by the substituent R. The CV curve of nitro derivative 5e demonstrated the minimum peak current. Considering that the current is a quantitative expression of how fast an electrochemical process is happening, compound 5e shows the lowest oxidation rate. In this case, two quasi-reversible reduction waves with lower E1/2ox compared to the other oligomers 5 were observed. Apparently, two nitrophenyl fragments are successively reduced in this process. A distinctive feature of the CV curves of compounds 5a and 5b was a more pronounced second oxidation wave. We speculated that the second oxidation of molecules 5 gives dicationic species (Scheme 5). Presumably, when R = H or OMe, structure 17 contributes the most to the resonance hybride, while in cases of molecules 5d,e with π-acceptor substituents, a dication of type 18 better describes the electron density distribution. The only exception is the trifluoromethyl derivative 5с, which shows the most positive oxidation potential and rate of the first oxidation as well as the lowest rate of reduction. However, the relatively small differences may simply be due to the different local solvation of the CF3 substituent.
![[1860-5397-19-49-11]](/bjoc/content/figures/1860-5397-19-49-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Cyclic voltammograms of oligomers 5.
Figure 11: Cyclic voltammograms of oligomers 5.
![[1860-5397-19-49-i5]](/bjoc/content/inline/1860-5397-19-49-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Possible ways of one- and two-electron oxidation of oligomers 5.
Scheme 5: Possible ways of one- and two-electron oxidation of oligomers 5.
Conclusion
Glaser–Hay homocoupling of 2-ethynyl-7-(arylethynyl)-1,8-bis(dimethylamino)naphthalenes yielded a series of previously unknown butadiynes 5 containing two fragments of arylethynyl substituted DMAN. The oligomers synthesized in this way are cross-conjugated systems, in which two independent conjugation pathways are realized: π-conjugation of DMAN fragments through a butadiyne linker and aryl–C≡C–DMAN conjugation paths. A comprehensive study by X-ray diffraction, NMR spectroscopy and cyclic voltammetry revealed that the “butadiyne pathway” is realized in cases where the aryl substituent is p-methoxyphenyl, phenyl or p-(trifluoromethyl)phenyl. Oligomers 5 bearing a π-acceptor para-nitro or para-cyano substituent on the aryl termini are A–π–D–π–D–π–A systems, which are characterized by the donor–acceptor conjugation pathway between the π-excessive DMAN residue and the π-deficient aryl ring. The effectiveness of this conjugation pathway was also confirmed by UV–vis spectroscopy data. Butadiynes 5 having electron-withdrawing CN and NO2 substituents in the aryl moieties demonstrated the longest wavelength absorption maxima in the series. The optical band gaps (Egopt), estimated from the onset point of the absorption spectra of 5, ranged within 2.39 eV (for phenyl and p-methoxyphenyl derivatives) to 2.09 eV (for p-nitrophenyl derivative). Thus, the HOMO–LUMO gap is significantly reduced by the introduction of the electron-withdrawing substituent, while the introduction of a donor substituent, e.g., a OMe group, does not change this value. On the other hand, compounds 5a–c (Ar = Ph, 4-MeOC6H4, 4-CF3C6H4) were red-shifted by 18–39 nm relatively to the corresponding monomers. Their absorption maxima were rather close to those of butadiyne 1 end-capped by two DMAN residues.
In doubly protonated tetrafluoroborate salts of the oligomers, conjugation between the aryl and naphthalene fragments becomes preferable, albeit diminished compared to bases 5. In the case of the 4-methoxyphenyl derivative, protonation results in the transformation of the D–π–D–π–D–π–D system into the D–π–A–π–A–π–D system.
Butadiynes 5 with terminal phenyl and p-methoxyphenyl groups demonstrated the lowest first oxidation potentials and the highest second oxidation rates, whereas p-nitrophenyl analogs showed the lowest oxidation rates and two quasi-reversible reduction waves with lower E1/2ox compared to other oligomers 5.
Upon prolonged exposure to a CHCl3/EtOH mixture, p-CF3C6H4-terminated butadiyne 5 gradually underwent demethylation/acid-catalyzed heterocyclization involving one of the dimethylamino groups and the adjacent C≡C bond of the butadiyne linker, forming the corresponding benzo[g]indole derivative.
Supporting Information
| Supporting Information File 1: Experimental section. | ||
| Format: PDF | Size: 5.5 MB | Download |
Acknowledgements
We thank Dr. Anna V. Tkachuk (Scientific and Educational Laboratory of Resonance Spectroscopy, Department of Natural and High Molecular Compounds Chemistry, Southern Federal University) for routine NMR measurements and Dr. Darya V. Spiridonova (Center for X-ray diffraction studies, St. Petersburg State University Research Park) for X-ray studies, Oussama Mammeri (Chemical analysis and materials research centre, St. Petersburg State University Research Park) for HRMS measurements, Dr. Pavel A. Knyazev (Southern Federal University) for CV experiments. Support for the X-ray instrument partially used in this study was also provided by the Center for collective use of scientific equipment of the North Caucasus Federal University financially supported by the Ministry of Education and Science of the Russian Federation (project identifier RF 2296.61321X0029, agreement No. 075-15-2021-687).
References
-
Forrest, S. R.; Thompson, M. E., Eds. Organic, Electronics, Optoelectronics. Chem. Rev. 2007, 107, 923–1386.
Return to citation in text: [1] [2] -
Miller, R. D.; Chandross, E. A., Eds. Materials for Electronics. Chem. Rev. 2010, 110, 1–574.
Return to citation in text: [1] [2] -
Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Chem. Rev. 2004, 104, 4971–5004. doi:10.1021/cr040084k
Return to citation in text: [1] -
Liu, Z.; Zhang, G.; Zhang, D. Acc. Chem. Res. 2018, 51, 1422–1432. doi:10.1021/acs.accounts.8b00069
Return to citation in text: [1] -
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Bunz, U. H. F. Chem. Rev. 2000, 100, 1605–1644. doi:10.1021/cr990257j
Return to citation in text: [1] -
Lee, J.-E.; Yang, J.; Kim, D. Faraday Discuss. 2012, 155, 277–288. doi:10.1039/c1fd00082a
Return to citation in text: [1] [2] -
Hisaki, I.; Hiroto, S.; Kim, K. S.; Noh, S. B.; Kim, D.; Shinokubo, H.; Osuka, A. Angew. Chem., Int. Ed. 2007, 46, 5125–5128. doi:10.1002/anie.200700550
Return to citation in text: [1] [2] -
Lee, S.; Chung, H.; Tokuji, S.; Yorimitsu, H.; Osuka, A.; Kim, D. Chem. Commun. 2014, 50, 2947–2950. doi:10.1039/c4cc00063c
Return to citation in text: [1] [2] -
Kuimova, M. K.; Balaz, M.; Anderson, H. L.; Ogilby, P. R. J. Am. Chem. Soc. 2009, 131, 7948–7949. doi:10.1021/ja901237s
Return to citation in text: [1] [2] -
Sakamoto, R.; Fukui, N.; Maeda, H.; Matsuoka, R.; Toyoda, R.; Nishihara, H. Adv. Mater. (Weinheim, Ger.) 2019, 31, 1804211. doi:10.1002/adma.201804211
Return to citation in text: [1] [2] -
Witulski, B.; Schweikert, T.; Schollmeyer, D.; Nemkovich, N. A. Chem. Commun. 2010, 46, 2953–2955. doi:10.1039/b919275a
Return to citation in text: [1] -
Doan, T.-H.; Talbi, I.; Lohier, J.-F.; Touil, S.; Alayrac, C.; Witulski, B. J. Mol. Struct. 2016, 1116, 127–134. doi:10.1016/j.molstruc.2016.03.026
Return to citation in text: [1] [2] -
Cataldo, F.; Ravagnan, L.; Cinquanta, E.; Castelli, I. E.; Manini, N.; Onida, G.; Milani, P. J. Phys. Chem. B 2010, 114, 14834–14841. doi:10.1021/jp104863v
Return to citation in text: [1] -
Filatova, E. A.; Tsybulin, S. V.; Rybin, D. A.; Ozeryanskii, V. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Borodkin, G. S. New J. Chem. 2022, 46, 1829–1838. doi:10.1039/d1nj05350g
Return to citation in text: [1] [2] [3] [4] -
Phelan, N. F.; Orchin, M. J. Chem. Educ. 1968, 45, 633–637. doi:10.1021/ed045p633
Return to citation in text: [1] -
Hosoya, H. Curr. Org. Chem. 2015, 19, 293–310. doi:10.2174/1385272819666141216231017
Return to citation in text: [1] [2] -
Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573
Return to citation in text: [1] [2] -
Gu, J.; Wu, W.; Stuyver, T.; Danovich, D.; Hoffmann, R.; Tsuji, Y.; Shaik, S. J. Am. Chem. Soc. 2019, 141, 6030–6047. doi:10.1021/jacs.9b01420
Return to citation in text: [1] [2] -
Hopf, H.; Sherburn, M. S., Eds. Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry; Wiley-VCH: Weinheim, Germany, 2016. doi:10.1002/9783527671182
Return to citation in text: [1] [2] -
Baghernejad, M.; Zhao, X.; Baruël Ørnsø, K.; Füeg, M.; Moreno-García, P.; Rudnev, A. V.; Kaliginedi, V.; Vesztergom, S.; Huang, C.; Hong, W.; Broekmann, P.; Wandlowski, T.; Thygesen, K. S.; Bryce, M. R. J. Am. Chem. Soc. 2014, 136, 17922–17925. doi:10.1021/ja510335z
Return to citation in text: [1] -
Andrews, D. Q.; Solomon, G. C.; Van Duyne, R. P.; Ratner, M. A. J. Am. Chem. Soc. 2008, 130, 17309–17319. doi:10.1021/ja804399q
Return to citation in text: [1] -
Chen, S.; Chen, G.; Ratner, M. A. J. Phys. Chem. Lett. 2018, 9, 2843–2847. doi:10.1021/acs.jpclett.8b01185
Return to citation in text: [1] -
Zhang, Y.; Ye, G.; Soni, S.; Qiu, X.; Krijger, T. L.; Jonkman, H. T.; Carlotti, M.; Sauter, E.; Zharnikov, M.; Chiechi, R. C. Chem. Sci. 2018, 9, 4414–4423. doi:10.1039/c8sc00165k
Return to citation in text: [1] -
Solomon, G. C.; Andrews, D. Q.; Goldsmith, R. H.; Hansen, T.; Wasielewski, M. R.; Van Duyne, R. P.; Ratner, M. A. J. Am. Chem. Soc. 2008, 130, 17301–17308. doi:10.1021/ja8044053
Return to citation in text: [1] -
Tykwinski, R. R.; Gubler, U.; Martin, R. E.; Diederich, F.; Bosshard, C.; Günter, P. J. Phys. Chem. B 1998, 102, 4451–4465. doi:10.1021/jp980829o
Return to citation in text: [1] -
Michinobu, T.; May, J. C.; Lim, J. H.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Gross, M.; Biaggio, I.; Diederich, F. Chem. Commun. 2005, 737–739. doi:10.1039/b417393g
Return to citation in text: [1] -
Zhao, Y.; Slepkov, A. D.; Akoto, C. O.; McDonald, R.; Hegmann, F. A.; Tykwinski, R. R. Chem. – Eur. J. 2005, 11, 321–329. doi:10.1002/chem.200400822
Return to citation in text: [1] -
Michinobu, T.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Frank, B.; Moonen, N. N. P.; Gross, M.; Diederich, F. Chem. – Eur. J. 2006, 12, 1889–1905. doi:10.1002/chem.200501113
Return to citation in text: [1] -
Payne, A. D.; Willis, A. C.; Sherburn, M. S. J. Am. Chem. Soc. 2005, 127, 12188–12189. doi:10.1021/ja053772
Return to citation in text: [1] -
Filatova, E. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Ermolenko, E. A.; Ozeryanskii, V. A.; Misharev, A. D. ChemistrySelect 2020, 5, 9932–9945. doi:10.1002/slct.202002745
Return to citation in text: [1] [2] [3] -
Dehu, C.; Meyers, F.; Bredas, J. L. J. Am. Chem. Soc. 1993, 115, 6198–6206. doi:10.1021/ja00067a039
Return to citation in text: [1] -
Takamizawa, S.; Takasaki, Y.; Sasaki, T.; Ozaki, N. Nat. Commun. 2018, 9, 3984. doi:10.1038/s41467-018-06431-7
Return to citation in text: [1] -
Ciofini, I.; Le Bahers, T.; Adamo, C.; Odobel, F.; Jacquemin, D. J. Phys. Chem. C 2012, 116, 11946–11955. doi:10.1021/jp3030667
Return to citation in text: [1] -
Stiegman, A. E.; Graham, E.; Perry, K. J.; Khundkar, L. R.; Cheng, L. T.; Perry, J. W. J. Am. Chem. Soc. 1991, 113, 7658–7666. doi:10.1021/ja00020a030
Return to citation in text: [1] -
Filatova, E. A.; Gulevskaya, A. V.; Pozharskii, A. F. Chem. Heterocycl. Compd. 2018, 54, 38–42. doi:10.1007/s10593-018-2227-9
Return to citation in text: [1] -
Shuzo, A.; Kunihiko, T.; Shin'ichi, N.; Kenichiro, N.; Kazuko, A.; Miwa, W. Bull. Chem. Soc. Jpn. 1995, 68, 2043–2051. doi:10.1246/bcsj.68.2043
Return to citation in text: [1] -
Stiegman, A. E.; Miskowski, V. M.; Perry, J. W.; Coulter, D. R. J. Am. Chem. Soc. 1987, 109, 5884–5886. doi:10.1021/ja00253a070
Return to citation in text: [1] -
Filatova, E. A.; Pozharskii, A. F.; Gulevskaya, A. V.; Ozeryanskii, V. A.; Tsybulin, S. V.; Filarowski, A. Eur. J. Org. Chem. 2019, 7128–7141. doi:10.1002/ejoc.201901292
Return to citation in text: [1] -
Pozharskii, A. F.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Degtyarev, A. V.; Kazheva, O. N.; Alexandrov, G. G.; Dyachenko, O. A. J. Org. Chem. 2003, 68, 10109–10122. doi:10.1021/jo035350t
Return to citation in text: [1]
| 39. | Filatova, E. A.; Pozharskii, A. F.; Gulevskaya, A. V.; Ozeryanskii, V. A.; Tsybulin, S. V.; Filarowski, A. Eur. J. Org. Chem. 2019, 7128–7141. doi:10.1002/ejoc.201901292 |
| 15. | Filatova, E. A.; Tsybulin, S. V.; Rybin, D. A.; Ozeryanskii, V. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Borodkin, G. S. New J. Chem. 2022, 46, 1829–1838. doi:10.1039/d1nj05350g |
| 40. | Pozharskii, A. F.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Degtyarev, A. V.; Kazheva, O. N.; Alexandrov, G. G.; Dyachenko, O. A. J. Org. Chem. 2003, 68, 10109–10122. doi:10.1021/jo035350t |
| 1. | Forrest, S. R.; Thompson, M. E., Eds. Organic, Electronics, Optoelectronics. Chem. Rev. 2007, 107, 923–1386. |
| 2. | Miller, R. D.; Chandross, E. A., Eds. Materials for Electronics. Chem. Rev. 2010, 110, 1–574. |
| 7. | Lee, J.-E.; Yang, J.; Kim, D. Faraday Discuss. 2012, 155, 277–288. doi:10.1039/c1fd00082a |
| 8. | Hisaki, I.; Hiroto, S.; Kim, K. S.; Noh, S. B.; Kim, D.; Shinokubo, H.; Osuka, A. Angew. Chem., Int. Ed. 2007, 46, 5125–5128. doi:10.1002/anie.200700550 |
| 9. | Lee, S.; Chung, H.; Tokuji, S.; Yorimitsu, H.; Osuka, A.; Kim, D. Chem. Commun. 2014, 50, 2947–2950. doi:10.1039/c4cc00063c |
| 30. | Payne, A. D.; Willis, A. C.; Sherburn, M. S. J. Am. Chem. Soc. 2005, 127, 12188–12189. doi:10.1021/ja053772 |
| 7. | Lee, J.-E.; Yang, J.; Kim, D. Faraday Discuss. 2012, 155, 277–288. doi:10.1039/c1fd00082a |
| 8. | Hisaki, I.; Hiroto, S.; Kim, K. S.; Noh, S. B.; Kim, D.; Shinokubo, H.; Osuka, A. Angew. Chem., Int. Ed. 2007, 46, 5125–5128. doi:10.1002/anie.200700550 |
| 9. | Lee, S.; Chung, H.; Tokuji, S.; Yorimitsu, H.; Osuka, A.; Kim, D. Chem. Commun. 2014, 50, 2947–2950. doi:10.1039/c4cc00063c |
| 10. | Kuimova, M. K.; Balaz, M.; Anderson, H. L.; Ogilby, P. R. J. Am. Chem. Soc. 2009, 131, 7948–7949. doi:10.1021/ja901237s |
| 11. | Sakamoto, R.; Fukui, N.; Maeda, H.; Matsuoka, R.; Toyoda, R.; Nishihara, H. Adv. Mater. (Weinheim, Ger.) 2019, 31, 1804211. doi:10.1002/adma.201804211 |
| 12. | Witulski, B.; Schweikert, T.; Schollmeyer, D.; Nemkovich, N. A. Chem. Commun. 2010, 46, 2953–2955. doi:10.1039/b919275a |
| 13. | Doan, T.-H.; Talbi, I.; Lohier, J.-F.; Touil, S.; Alayrac, C.; Witulski, B. J. Mol. Struct. 2016, 1116, 127–134. doi:10.1016/j.molstruc.2016.03.026 |
| 14. | Cataldo, F.; Ravagnan, L.; Cinquanta, E.; Castelli, I. E.; Manini, N.; Onida, G.; Milani, P. J. Phys. Chem. B 2010, 114, 14834–14841. doi:10.1021/jp104863v |
| 15. | Filatova, E. A.; Tsybulin, S. V.; Rybin, D. A.; Ozeryanskii, V. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Borodkin, G. S. New J. Chem. 2022, 46, 1829–1838. doi:10.1039/d1nj05350g |
| 21. | Baghernejad, M.; Zhao, X.; Baruël Ørnsø, K.; Füeg, M.; Moreno-García, P.; Rudnev, A. V.; Kaliginedi, V.; Vesztergom, S.; Huang, C.; Hong, W.; Broekmann, P.; Wandlowski, T.; Thygesen, K. S.; Bryce, M. R. J. Am. Chem. Soc. 2014, 136, 17922–17925. doi:10.1021/ja510335z |
| 22. | Andrews, D. Q.; Solomon, G. C.; Van Duyne, R. P.; Ratner, M. A. J. Am. Chem. Soc. 2008, 130, 17309–17319. doi:10.1021/ja804399q |
| 23. | Chen, S.; Chen, G.; Ratner, M. A. J. Phys. Chem. Lett. 2018, 9, 2843–2847. doi:10.1021/acs.jpclett.8b01185 |
| 24. | Zhang, Y.; Ye, G.; Soni, S.; Qiu, X.; Krijger, T. L.; Jonkman, H. T.; Carlotti, M.; Sauter, E.; Zharnikov, M.; Chiechi, R. C. Chem. Sci. 2018, 9, 4414–4423. doi:10.1039/c8sc00165k |
| 25. | Solomon, G. C.; Andrews, D. Q.; Goldsmith, R. H.; Hansen, T.; Wasielewski, M. R.; Van Duyne, R. P.; Ratner, M. A. J. Am. Chem. Soc. 2008, 130, 17301–17308. doi:10.1021/ja8044053 |
| 1. | Forrest, S. R.; Thompson, M. E., Eds. Organic, Electronics, Optoelectronics. Chem. Rev. 2007, 107, 923–1386. |
| 2. | Miller, R. D.; Chandross, E. A., Eds. Materials for Electronics. Chem. Rev. 2010, 110, 1–574. |
| 3. | Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Chem. Rev. 2004, 104, 4971–5004. doi:10.1021/cr040084k |
| 4. | Liu, Z.; Zhang, G.; Zhang, D. Acc. Chem. Res. 2018, 51, 1422–1432. doi:10.1021/acs.accounts.8b00069 |
| 5. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 26. | Tykwinski, R. R.; Gubler, U.; Martin, R. E.; Diederich, F.; Bosshard, C.; Günter, P. J. Phys. Chem. B 1998, 102, 4451–4465. doi:10.1021/jp980829o |
| 27. | Michinobu, T.; May, J. C.; Lim, J. H.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Gross, M.; Biaggio, I.; Diederich, F. Chem. Commun. 2005, 737–739. doi:10.1039/b417393g |
| 28. | Zhao, Y.; Slepkov, A. D.; Akoto, C. O.; McDonald, R.; Hegmann, F. A.; Tykwinski, R. R. Chem. – Eur. J. 2005, 11, 321–329. doi:10.1002/chem.200400822 |
| 29. | Michinobu, T.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Frank, B.; Moonen, N. N. P.; Gross, M.; Diederich, F. Chem. – Eur. J. 2006, 12, 1889–1905. doi:10.1002/chem.200501113 |
| 15. | Filatova, E. A.; Tsybulin, S. V.; Rybin, D. A.; Ozeryanskii, V. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Borodkin, G. S. New J. Chem. 2022, 46, 1829–1838. doi:10.1039/d1nj05350g |
| 17. | Hosoya, H. Curr. Org. Chem. 2015, 19, 293–310. doi:10.2174/1385272819666141216231017 |
| 18. | Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573 |
| 19. | Gu, J.; Wu, W.; Stuyver, T.; Danovich, D.; Hoffmann, R.; Tsuji, Y.; Shaik, S. J. Am. Chem. Soc. 2019, 141, 6030–6047. doi:10.1021/jacs.9b01420 |
| 20. | Hopf, H.; Sherburn, M. S., Eds. Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry; Wiley-VCH: Weinheim, Germany, 2016. doi:10.1002/9783527671182 |
| 11. | Sakamoto, R.; Fukui, N.; Maeda, H.; Matsuoka, R.; Toyoda, R.; Nishihara, H. Adv. Mater. (Weinheim, Ger.) 2019, 31, 1804211. doi:10.1002/adma.201804211 |
| 17. | Hosoya, H. Curr. Org. Chem. 2015, 19, 293–310. doi:10.2174/1385272819666141216231017 |
| 18. | Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573 |
| 19. | Gu, J.; Wu, W.; Stuyver, T.; Danovich, D.; Hoffmann, R.; Tsuji, Y.; Shaik, S. J. Am. Chem. Soc. 2019, 141, 6030–6047. doi:10.1021/jacs.9b01420 |
| 20. | Hopf, H.; Sherburn, M. S., Eds. Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry; Wiley-VCH: Weinheim, Germany, 2016. doi:10.1002/9783527671182 |
| 13. | Doan, T.-H.; Talbi, I.; Lohier, J.-F.; Touil, S.; Alayrac, C.; Witulski, B. J. Mol. Struct. 2016, 1116, 127–134. doi:10.1016/j.molstruc.2016.03.026 |
| 10. | Kuimova, M. K.; Balaz, M.; Anderson, H. L.; Ogilby, P. R. J. Am. Chem. Soc. 2009, 131, 7948–7949. doi:10.1021/ja901237s |
| 16. | Phelan, N. F.; Orchin, M. J. Chem. Educ. 1968, 45, 633–637. doi:10.1021/ed045p633 |
| 32. | Dehu, C.; Meyers, F.; Bredas, J. L. J. Am. Chem. Soc. 1993, 115, 6198–6206. doi:10.1021/ja00067a039 |
| 31. | Filatova, E. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Ermolenko, E. A.; Ozeryanskii, V. A.; Misharev, A. D. ChemistrySelect 2020, 5, 9932–9945. doi:10.1002/slct.202002745 |
| 31. | Filatova, E. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Ermolenko, E. A.; Ozeryanskii, V. A.; Misharev, A. D. ChemistrySelect 2020, 5, 9932–9945. doi:10.1002/slct.202002745 |
| 37. | Shuzo, A.; Kunihiko, T.; Shin'ichi, N.; Kenichiro, N.; Kazuko, A.; Miwa, W. Bull. Chem. Soc. Jpn. 1995, 68, 2043–2051. doi:10.1246/bcsj.68.2043 |
| 38. | Stiegman, A. E.; Miskowski, V. M.; Perry, J. W.; Coulter, D. R. J. Am. Chem. Soc. 1987, 109, 5884–5886. doi:10.1021/ja00253a070 |
| 35. | Stiegman, A. E.; Graham, E.; Perry, K. J.; Khundkar, L. R.; Cheng, L. T.; Perry, J. W. J. Am. Chem. Soc. 1991, 113, 7658–7666. doi:10.1021/ja00020a030 |
| 36. | Filatova, E. A.; Gulevskaya, A. V.; Pozharskii, A. F. Chem. Heterocycl. Compd. 2018, 54, 38–42. doi:10.1007/s10593-018-2227-9 |
| 15. | Filatova, E. A.; Tsybulin, S. V.; Rybin, D. A.; Ozeryanskii, V. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Borodkin, G. S. New J. Chem. 2022, 46, 1829–1838. doi:10.1039/d1nj05350g |
| 34. | Ciofini, I.; Le Bahers, T.; Adamo, C.; Odobel, F.; Jacquemin, D. J. Phys. Chem. C 2012, 116, 11946–11955. doi:10.1021/jp3030667 |
| 33. | Takamizawa, S.; Takasaki, Y.; Sasaki, T.; Ozaki, N. Nat. Commun. 2018, 9, 3984. doi:10.1038/s41467-018-06431-7 |
| 31. | Filatova, E. A.; Gulevskaya, A. V.; Pozharskii, A. F.; Ermolenko, E. A.; Ozeryanskii, V. A.; Misharev, A. D. ChemistrySelect 2020, 5, 9932–9945. doi:10.1002/slct.202002745 |
© 2023 Tsybulin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.