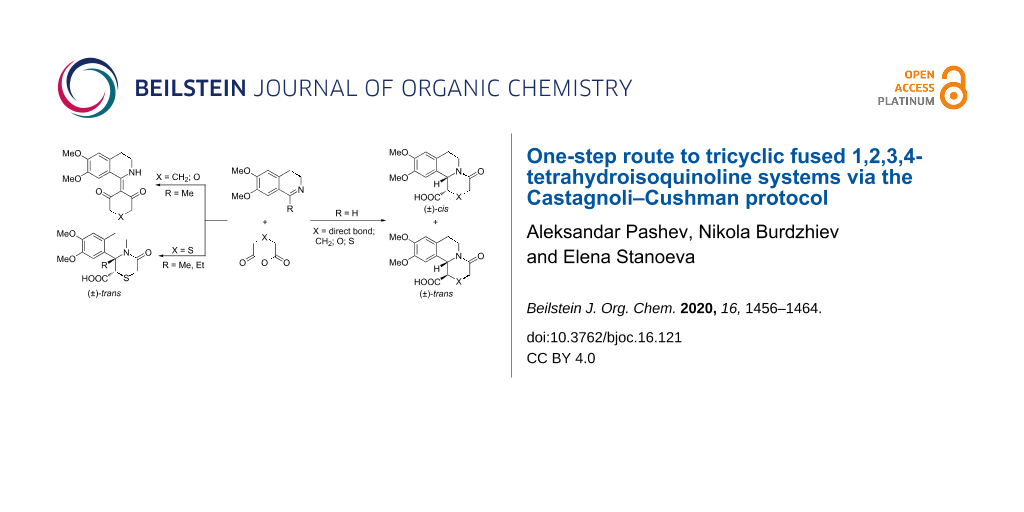
Abstract
The Castagnoli–Cushman reaction of 3,4-dihydroisoquinolines with glutaric anhydride, its oxygen and sulfur analogues was investigated as a one-step approach to the benzo[a]quinolizidine system and its heterocyclic analogs. An extension towards the pyrrolo[2,1-a]isoquinoline system was achieved with the use of succinic anhydride. The results are evidence of an unexplored method for the access of the aforementioned tricyclic annelated systems incorporating a bridgehead nitrogen atom. The structures and relative configurations of the new compounds were established by means of 1D and 2D NMR techniques. The reactions between 1-methyldihydroisoquinoline and glutaric, diglycolic and succinic anhydrides yielded unexpected isoquinoline derivatives containing an exocyclic double bond. The compounds prepared bear the potential to become building blocks for future synthetic bioactive molecules.

Graphical Abstract
Introduction
The benzo[a]quinolizidine ring system is an important heterocyclic framework found in natural products and prospective pharmaceuticals [1]. This heterocycle is present in several alkaloids such as emetine (1) and related compounds, that exhibit biological activities such as glucosidase inhibition, anti-amoebic properties as well as activity against breast cancer cell lines [2]. Notable examples of synthetic compounds include the dipeptidyl peptidase IV inhibitor carmegliptin (2, DPP IV) with potential for the treatment of type-II diabetes [3,4]. A comparative study on novel classes of anticancer drugs identified benzo[a]quinolizines 3 and 4 (Figure 1) to be useful for a specific inhibition of heat shock response in cancer cells, which strongly enhances the treatment by sensitizing cancer cells to anticancer drugs [5]. The presence of such structural pattern has driven the development of various approaches for its obtaining – based either on isolation from naturally occurring sources or through multistep synthetic routes [6].
![[1860-5397-16-121-1]](/bjoc/content/figures/1860-5397-16-121-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Compounds comprising a benzo[a]quinolizidine ring system.
Figure 1: Compounds comprising a benzo[a]quinolizidine ring system.
The pyrrolo[2,1-a]isoquinoline skeleton is incorporated in a large group of natural compounds such as crispines, trolline, and lamellarins, which are interesting due to their anticancer, antiviral, and antibacterial activities [7,8]. In the light of this broad array of biological activities, the development of novel methods for the construction of these heteropolycycles is of great significance to both organic and medicinal chemistry.
The Castagnoli–Cushman reaction (CCR) between cyclic enolizable anhydrides (such as succinic (5) [9], glutaric (6) [10], and the corresponding oxygen (7) [11], sulfur (8) [11], and nitrogen analogs 9 [12], and homophthalic anhydride (10) [13,14]) and imines 11, 12 offers a route to substituted lactam molecules 13 and 14 [13-19]. Among them, the reactions of homophthalic anhydride (10) with Schiff bases 11 or cyclic imines 12 have been examined in detail (Scheme 1) [13,14,20,21].
![[1860-5397-16-121-i1]](/bjoc/content/inline/1860-5397-16-121-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions between enolizable anhydrides and imines.
Scheme 1: Reactions between enolizable anhydrides and imines.
The mechanism of the reaction is still under debate with two prevailing versions in the literature (Scheme 2) [16,17,22-24]. The first reaction pathway includes the formation of an N-acyliminium ion 15, followed by a ring closure through an enolate ion 16. The other mechanistic proposal features a stepwise Mannich-type reaction of the enolized anhydride 17 to the imine component and a subsequent N-acylation reaction to form the lactam target product [24]. A respective Mannich-type intermediate has been recently isolated and subsequently converted into the target lactam product [24]. It is also suggested that the imine substrate structure and the anhydride’s ability to undergo enolization have a profound influence on the reaction course [16,23].
![[1860-5397-16-121-i2]](/bjoc/content/inline/1860-5397-16-121-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanistic pathways for the reaction between cyclic anhydrides and imines.
Scheme 2: Mechanistic pathways for the reaction between cyclic anhydrides and imines.
While the reactions of homophthalic anhydride (10) and various cyclic imines 12 have been extensively explored [13,21,24,25], there are only few reports featuring reactions between monocyclic anhydrides 5–9 and cyclic imines 12 in the literature [12,25-29]. Thus, it was shown that glutaconic anhydride reacts with 1-methyl-3,4-dihydroisoquinoline and with 1-chloroisoquinoline to give 11b-methylbenzo[a]quinolizin-4-one [28] and benzo[a]quinolizin-4-one [27], respectively, in moderate yields. Attempts by Krasavin et al. showed that isoquinoline itself demonstrated modest reactivity and diastereoselectivity in reactions with thiodiacetic anhydride (8) [12]. It can be concluded that due to their stability the aromatic heterocycles have a low tendency to undergo reactions with cyclic anhydrides, resulting in low to moderate overall yields of the target products [12,27]. Further research by Krasavin et al. on the reactivity of sterically hindered indolenines in reactions with anhydrides 6–9, did not give the expected fused δ-lactams [26].
The purpose of the present investigation was to explore the much less studied reaction between monocyclic anhydrides 5–8 and cyclic imines (such as 6,7-dimethoxy-3,4-dihydroisoquinoline (18) and its 1-alkyl derivatives 19 and 20) as a potential one-step route towards substituted benzo[a]quinolizidinones and pyrrolo[2,1-a]isoquinolinones. This reaction also would allow the construction of bioisosteric oxygen and sulfur derivatives of the target heterocyclic framework.
Results and Discussion
Our synthetic strategy relied on the possibility to form ring C of the target ring system using the Castagnoli–Cushman reaction between 6,7-dimethoxy-3,4-dihydroisoquinoline (18) and its 1-substituted derivatives 19 and 20, and the anhydrides 5–8. This approach also could allow the construction of bioisosteric O- and S-analogs of the target systems as it is shown by the retrosynthetic analysis (Scheme 3). For the present studies the employed starting imines 18–20, were prepared by a Bischler–Napieralski condensation, using procedures available in the literature [30,31].
![[1860-5397-16-121-i3]](/bjoc/content/inline/1860-5397-16-121-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Retrosynthetic analysis of the target compounds.
Scheme 3: Retrosynthetic analysis of the target compounds.
Reactions of 6,7-dimethoxy-3,4-dihydroisoquinoline (18) with anhydrides 5–8
The reaction between 18 and anhydrides 5–8 was conducted in xylene under inert atmosphere at 120 °C (Scheme 4). Previous knowledge on the reaction between monocyclic anhydrides 5, 6 and 7 and Schiff bases indicated the necessity of elevated temperatures for conducting the reaction as the yields of the target products increased with the temperature [32-34]. The anhydride was taken in slight molar excess in analogy to our previous work [21].
![[1860-5397-16-121-i4]](/bjoc/content/inline/1860-5397-16-121-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of 6,7-dimethoxy-3,4-dihydroisoquinoline (18) with anhydrides 5–8. Reagents and conditions: xylene, inert atmosphere, 120 °C, 6 h.
Scheme 4: Reaction of 6,7-dimethoxy-3,4-dihydroisoquinoline (18) with anhydrides 5–8. Reagents and conditions...
The reaction mixtures were heated for 6 h, until the imine 18 reacted completely. In the course of the reaction two stereocenters were formed at C-10b for 21 and C-11b for 22–24, respectively, and C–1. It should be noted that the compounds described here are racemic. After removal of xylene the 1H NMR spectrum of the residue was taken to give the diastereomeric cis:trans ratio, which in the reaction mixtures was 1:1. The obtained mixtures of diastereomers 21–24 were separated by column chromatography to give the individual diastereomers. The acid (−)-trans-22 and its methyl ester are known compounds, which were prepared by an independent method [35].
NOESY experiments were used to examine the stereochemical assignments in compounds 21–24. The cis diastereomers 22–24 showed distinct NOE cross-signals between protons H-1 and H-11b, and for cis-21 – between H-1 and H-10b. No such NOE peaks were identified in the spectra of the trans isomers (Figure 2). Further, the assignment of the relative configuration of the products 22–24 was supported by the vicinal coupling constants 3J1,11b for H-1 and H-11b. The 1H NMR spectra of the trans diastereomers were expected to display a larger coupling constant 3J1,11b in comparison with the spectra of the corresponding cis analogs [36]. A similar difference was observed in the spectra of the diastereomeric dibenzo[a,g]quinolizidinone compounds as well [14,37-39].
![[1860-5397-16-121-2]](/bjoc/content/figures/1860-5397-16-121-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Representative NOE interactions in cis and trans-21–24 (only one enantiomer is shown).
Figure 2: Representative NOE interactions in cis and trans-21–24 (only one enantiomer is shown).
As it can be seen from Table 1, the spectra of cis-22–24 displayed 3J1,11b within the narrow interval of 4.0–4.2 Hz, which could be attributed to a relatively more rigid conformation in the cis isomers. The spectra of the trans-22–24 compounds exhibited 3J1,11b in a broader interval of 5.7–10.3 Hz, which could be explained by a higher conformational flexibility of the trans isomers. Similar values of 3J1,11b were observed in the spectra of the known (−)-trans-22 [35] and the closely related structural analogs trans-1-ethylbenzo[a]quinolizidin-4-ones, previously described by Amat et al. [40]. The 1H NMR spectra of trans-21 and cis-21 displayed very close values for 3J1,10b, i.e., 6.9 Hz for trans-21, and 6.4 Hz for cis-21, which hindered the assignment of the relative configuration based solely upon the 3J values. However, taking into account the above-mentioned NOESY experiments, the relative configuration of compound 21 displaying 3J1,10b = 6.4 Hz was assigned as cis, and the configuration of the compound with 3J1,10b = 6.9 Hz as the trans isomer (Table 1).
Table 1: NMR data and stereochemical assignment of cis and trans-21–24.
|
H–1,
δ ppm |
H-11ba,
δ ppm |
3J1,11ba,
Hz |
|
| cis-21 | 3.90–4.04 m | 4.86 d | 6.4 Hz |
| trans-21 | 3.48 t | 4.97 d | 6.9 Hz |
| cis-22 | 4.64 m | 4.84 d | 4.2 Hz |
| trans-22 | 3.10 ddd | 4.93 d | 7.1 Hz |
| cis-23 | 4.96 d | 5.16 d | 4.2 Hz |
| trans-23 | 5.03 d | 4.73 d | 5.7 Hz |
| cis-24 | 4.54 d | 5.08 d | 4.0 Hz |
| trans-24 | 3.55 d | 5.05 d | 10.3 Hz |
aFor compounds cis and trans-21 the values for H-10b and 3J1,10b are given.
As it is known, the benzo[a]quinolizidine system can exist as interconvertible conformers due to the presence of a stereochemically mobile nitrogen atom [41]. Numbers of studies suggested that the preferred conformation of benzo[a]quinolizidines depended strongly on the type of substituents, the presence of stereocenters and their configuration [36,41-46]. As for the 4-oxobenzo[a]quinolizidine derivatives, it should be noted that due to the planar amide fragment the interconversion of the different conformers could be largely suppressed. The X-ray analysis of the above-mentioned (−)-trans-22 [35] and trans-1-ethylbenzo[a]quinolizin-4-ones [40] showed a distortion of the B and C rings, while the substituent at C-1 acquired a spatial orientation with a smaller steric repulsion with H-11. Based on the data outlaid above the assignment of the preferred conformation of the acids trans- and cis- 21–24 posed significant challenge.
Reactions of 1-alkyl-3,4-dihydroisoquinolines 19 and 20 with anhydrides 5–8
The reactions of 1-methyl and 1-ethyl-6,7-dimethoxy-3,4-dihydroisoquinoline 19 and 20 with anhydrides 5–8 were investigated under the same reaction conditions as the reactions of imine 18. When glutaric (6) and diglycolic anhydride (7) were subjected to the reaction with 19, the reaction yielded the unexpected products 25 and 26, respectively, containing an exocyclic double bond to the tetrahydroisoquinoline moiety (Scheme 5). The reaction with succinic anhydride (5) furnished the tetrahydroisoquinoline 27 with an open-chain acid substituent to the exocyclic double bond. Unlike 1-methyldihydroisoquinoline (19), the 1-ethyl derivative 20 did not react at all with the anhydrides 5–7.
![[1860-5397-16-121-i5]](/bjoc/content/inline/1860-5397-16-121-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of 1-methyl-3,4-dihydroisoquinoline (19) with anhydrides 5–7. Reagents and conditions: xylene, inert atmosphere, 120 °C, 6 h.
Scheme 5: Reaction of 1-methyl-3,4-dihydroisoquinoline (19) with anhydrides 5–7. Reagents and conditions: xyl...
The structures of compounds 25 and 26 were confirmed by 2D NMR spectroscopy and singe-crystal X-ray analysis (Figure 3).
![[1860-5397-16-121-3]](/bjoc/content/figures/1860-5397-16-121-3.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of products 25 and 26.
Figure 3: X-ray crystal structure of products 25 and 26.
The piperidine moiety is planar to a significant extent due to the combined influence of the aromatic ring and the exocyclic C–C double bond. The geometry of the nitrogen atom is also planar most likely because of conjugation with the exocyclic C–C double bond and the C=O groups. The nitrogen planarity is probably also caused by a hydrogen bond between the NH group and the neighboring carbonyl oxygen atom. The structure of 27 was proven by a combination of 1D and 2D NMR spectra, and IR spectroscopy.
The reactivity of thiodiacetic anhydride 8 towards the imines 19 and 20 was different to that of the anhydrides 5–7. The targeted 11b-methyl and 11b-ethyl-4-oxo-2-thiabenzo[a]quinolizidinones 28 and 29, respectively, were formed as single diastereomers (Scheme 6). The 1-ethyldihydroisoquinoline 20 showed a pronounced lower reactivity to anhydride 8, with a 30% yield of 29 as compared to 28 (71%), probably due to a steric hindrance.
![[1860-5397-16-121-i6]](/bjoc/content/inline/1860-5397-16-121-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactions of 1-alkyl-3,4-dihydroisoquinolines 19 and 20 with anhydride 8. Reagents and conditions: xylene, inert atmosphere, 120 °C, 6 h.
Scheme 6: Reactions of 1-alkyl-3,4-dihydroisoquinolines 19 and 20 with anhydride 8. Reagents and conditions: ...
The relative configuration of 28 and 29 was established using 2D ROESY NMR spectroscopy. The spectra showed cross peaks between the protons from the cis-oriented alkyl-C-11b and H-1, which unequivocally proved the trans relative configuration of the substituents at C-1 and C-11b (Figure 4).
![[1860-5397-16-121-4]](/bjoc/content/figures/1860-5397-16-121-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Representative NOE interactions in 28 and 29.
Figure 4: Representative NOE interactions in 28 and 29.
1-Alkyl-3,4-dihydroisoquinolines have been reported to react as C-nucleophiles or 1,3-C,N-ambident nucleophiles with electrophilic species [47-50].
Thus the formation of compounds 25–27 could be rationalized as including an initial nucleophilic attack of the enamine tautomer 19a of 3,4-dihydroisoquinoline 19 to the carbonyl carbon atoms of the anhydrides 5–7 followed by an anhydride ring opening. In the case of anhydrides 6 and 7, a 6-exo-trig ring-closure reaction of the tautomer form 30a leads to the observed products 25 and 26, according to the plausible mechanistic suggestion presented in Scheme 7.
![[1860-5397-16-121-i7]](/bjoc/content/inline/1860-5397-16-121-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Suggested mechanism for the formation of products 25–27.
Scheme 7: Suggested mechanism for the formation of products 25–27.
In the case of compound 27 (X = direct bond), the reaction could be characterized as a 5-exo-trig cyclization, which is favored as long as the Bürgi–Dunitz angle is achievable [51]. It could be concluded that the reaction does not proceed further probably because the shorter side chain makes difficult the achievement of the required angle of approach of the nucleophile.
The observed different reactivity of the anhydrides 5–7 towards 1-methyl-3,4-dihydroisoquinoline 19 needs additional explanation. Work in this field based on the theoretical treatment of the possible stabilities of the intermediates and the hypothetic transition states of the steps is in progress.
Conclusion
The scope of the Castagnoli–Cushman reaction was enlarged by the use of 3,4-dihydroisoquinolines and succinic, glutaric, diglycolic, and thiodiacetic anhydrides. The substituted benzo[a]quinolizidine products and their heterocyclic analogs as well as 3-oxopyrrolo[2,1-a]isoquinolin-1-carboxylic acids were obtained as a 1:1 mixture of diastereomers with varying yields when 3,4-dihydroisoquinoline was used. The reaction of 1-methyl and 1-ethyldihydroisoquinoline with thiodiacetic anhydride afforded the expected angularly substituted trans-2-thiabenzo[a]quinolizidinones. On the contrary, the glutaric, succinic, and diglycolic anhydrides reacted with 1-methyldihydroisoquinoline to give isoquinoline products with an exocyclic double bond. This research demonstrated once more the versatility of 3,4-dihydroisoquinolines as synthons for the preparation of fused polyheterocycles containing a bridgehead nitrogen atom.
Experimental
General
Melting points were taken on an automated melting point apparatus SRS EZ-Melt MPA120 at a ramp rate of 1 °C/min and are uncorrected. IR spectra were recorded on a Thermo Fischer Nicolet iS50 FT-IR instrument. Spectroscopic grade KBr was used in the sample preparation without further purification. 1H NMR spectra (500 MHz) and 13C NMR spectra (125 MHz) were obtained on a Bruker Avance III HD 500 MHz spectrometer. The chemical shifts are given in parts per million (δ) for the spectra in DMSO-d6 relative to the residual solvent peak [52]. Coupling constants (J) are reported in Hz. Assignments were made by using a combination of 1D and 2D spectra (COSY, HSQC and HMBC). Thin-layer chromatography (TLC) was performed on Merck 1.05554 silica gel 60F254 aluminum plates. Chromatographic filtration and column chromatography were carried out using Merck Kiesegel 60 (0.060–0.200 mm). High-resolution mass spectra (HRMS) were acquired on a Thermo Scientific, model Q-Exactive high resolution LC–MS/MS with a resolution of up to 150000FWHM at m/z 200.
X-ray structure determination: Colorless single crystals of compounds 25 and 26 were obtained from DMSO/H2O. Diffraction data were collected at 300 K the by v-scan technique, on a Mitigen Micromount and automatically centered on a Bruker SMART X2S Benchtop Crystallographic system. Intensity measurements were performed using monochromatized Mo Kα radiation (λ = 0.71073 Å) from a sealed microfocus tube and BREEZE CCD detector. The APEX 2 v.2014.11.0 software was used for all data processing [53]. The structure was solved and refined with SHELX [54] programs, ShelXT and ShelXL by using OLEX 2 software [55]. Hydrogen atoms were placed at idealized positions and refined. Crystallographic data (excluding structure factors) for the structural analysis were deposited with the Cambridge Crystallographic Data Centre, CCDC 1963825 and 1963826, respectively. Copy of this information can be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK. Fax: +44 1223 336–033, e-mail: deposit@ccdc.cam.ac.uk, or http://www.ccdc.cam.ac.uk.
General procedure for preparation of (±)-trans and cis-21–24
The starting 6,7-dimethoxy-3,4-dihydroisoquinolinium perchlorate (1.2 mmol, 0.35 g) was dissolved in water (50 mL) and the resulting suspension was treated with solid sodium hydroxide until pH 10. The resulting mixture was extracted with dichloromethane (3 × 10 mL) and the organic phase was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The obtained free base (0.21 g, 92%, 1.1 mmol) was dissolved in dry xylene (2.5 mL) and the corresponding anhydride (1.4 mmol) was added. The reaction mixture was heated at 120 °C under inert atmosphere for 6 hours. TLC was used to monitor the course of the reaction. Upon completion, the solvent was evaporated under reduced pressure and the resulting crude products were separated and purified by means of column chromatography. The ratio of the cis:trans diastereomers was obtained from the 1H NMR spectra of the crude reaction mixture.
General procedure for preparation of 25–28
The corresponding 1-methyl or 1-ethyl-3,4-dihydroisoquinoline (19, 1 mmol, 0.205 g; 20, 1 mmol, 0.219 g) was dissolved in dry xylene (2 mL) and the corresponding anhydride (1.5 mmol) was added. The reaction mixture was heated under inert atmosphere for 6 hours. TLC was used to monitor the course of the reaction. After completion, the solvent was evaporated under reduced pressure and the resulting crude products were isolated and purified by means of column chromatography, followed by recrystallization.
Supporting Information
| Supporting Information File 1: Experimental procedures for compounds 21–29 and their spectroscopic and analytic data. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Garrido Montalban, A. Quinolines and Isoquinolines. Heterocycles in Natural Product Synthesis; Wiley-VCH: Weinheim, Germany, 2011; pp 299–339. doi:10.1002/9783527634880.ch9
Return to citation in text: [1] -
Dwek, R. A.; Butters, T. D.; Platt, F. M.; Zitzmann, N. Nat. Rev. Drug Discovery 2002, 1, 65–75. doi:10.1038/nrd708
Return to citation in text: [1] -
Boehringer, M.; Fischer, H.; Hennig, M.; Hunziker, D.; Huwyler, J.; Kuhn, B.; Loeffler, B. M.; Luebbers, T.; Mattei, P.; Narquizian, R.; Sebokova, E.; Sprecher, U.; Wessel, H. P. Bioorg. Med. Chem. Lett. 2010, 20, 1106–1108. doi:10.1016/j.bmcl.2009.12.025
Return to citation in text: [1] -
Mattei, P.; Boehringer, M.; Di Giorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U. Bioorg. Med. Chem. Lett. 2010, 20, 1109–1113. doi:10.1016/j.bmcl.2009.12.024
Return to citation in text: [1] -
Zaarur, N.; Gabai, V. L.; Porco, J. A., Jr.; Calderwood, S.; Sherman, M. Y. Cancer Res. 2006, 66, 1783–1791. doi:10.1158/0008-5472.can-05-3692
Return to citation in text: [1] -
Pashev, A. S.; Burdzhiev, N. T.; Stanoeva, E. R. Org. Prep. Proced. Int. 2016, 48, 425–467. doi:10.1080/00304948.2016.1234820
Return to citation in text: [1] -
Matveeva, M. D.; Purgatorio, R.; Voskressensky, L. G.; Altomare, C. D. Future Med. Chem. 2019, 11, 2735–2755. doi:10.4155/fmc-2019-0136
Return to citation in text: [1] -
Pässler, U.; Knölker, H.-J. The Pyrrolo[2,1-a]isoquinoline Alkaloids. Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, Netherlands, 2011; Vol. 70, pp 79–151. doi:10.1016/b978-0-12-391426-2.00002-5
Return to citation in text: [1] -
Castagnoli, N. J. Org. Chem. 1969, 34, 3187–3189. doi:10.1021/jo01262a081
Return to citation in text: [1] -
Cushman, M.; Castagnoli, N. J. Org. Chem. 1973, 38, 440–448. doi:10.1021/jo00943a007
Return to citation in text: [1] -
Shetty, B. V.; McFadden, A.; Hofer, P. Analgesic 4-carboxy-pyrrolidin-2-one compound. U.S. Patent US4,476,311, Oct 9, 1984.
Return to citation in text: [1] [2] -
Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014
Return to citation in text: [1] [2] [3] [4] -
Haimova, M. A.; Mollov, N. M.; Ivanova, S. C.; Dimitrova, A. I.; Ognyanov, V. I. Tetrahedron 1977, 33, 331–336. doi:10.1016/0040-4020(77)80114-2
Return to citation in text: [1] [2] [3] [4] -
Cushman, M.; Gentry, J.; Dekow, F. W. J. Org. Chem. 1977, 42, 1111–1116. doi:10.1021/jo00427a001
Return to citation in text: [1] [2] [3] [4] -
Shaw, J. T. Nat. Prod. Rep. 2009, 26, 11–26. doi:10.1039/b814468k
Return to citation in text: [1] -
Ng, P. Y.; Masse, C. E.; Shaw, J. T. Org. Lett. 2006, 8, 3999–4002. doi:10.1021/ol061473z
Return to citation in text: [1] [2] [3] -
Di Maso, M. J.; Snyder, K. M.; De Souza Fernandes, F.; Pattawong, O.; Tan, D. Q.; Fettinger, J. C.; Cheong, P. H.-Y.; Shaw, J. T. Chem. – Eur. J. 2016, 22, 4794–4801. doi:10.1002/chem.201504424
Return to citation in text: [1] [2] -
Tan, D. Q.; Younai, A.; Pattawong, O.; Fettinger, J. C.; Cheong, P. H.-Y.; Shaw, J. T. Org. Lett. 2013, 15, 5126–5129. doi:10.1021/ol402554n
Return to citation in text: [1] -
Pattawong, O.; Tan, D. Q.; Fettinger, J. C.; Shaw, J. T.; Cheong, P. H.-Y. Org. Lett. 2013, 15, 5130–5133. doi:10.1021/ol402561q
Return to citation in text: [1] -
Georgieva, A.; Stanoeva, E.; Spassov, S.; Macicek, J.; Angelova, O.; Haimova, M.; De Kimpe, N. Tetrahedron 1991, 47, 3375–3388. doi:10.1016/s0040-4020(01)86402-4
Return to citation in text: [1] -
Stanoeva, E.; Georgieva, A.; Avramova, S.; Burdzhiev, N.; Lázár, L. J. Heterocycl. Chem. 2015, 52, 130–135. doi:10.1002/jhet.1965
Return to citation in text: [1] [2] [3] -
Cushman, M.; Madaj, E. J. J. Org. Chem. 1987, 52, 907–915. doi:10.1021/jo00381a033
Return to citation in text: [1] -
Tang, Y.; Fettinger, J. C.; Shaw, J. T. Org. Lett. 2009, 11, 3802–3805. doi:10.1021/ol901018k
Return to citation in text: [1] [2] -
Bakulina, O.; Ivanov, A.; Suslonov, V.; Dar’in, D.; Krasavin, M. Beilstein J. Org. Chem. 2017, 13, 1413–1424. doi:10.3762/bjoc.13.138
Return to citation in text: [1] [2] [3] [4] -
González-López, M.; Shaw, J. T. Chem. Rev. 2009, 109, 164–189. doi:10.1021/cr8002714
Return to citation in text: [1] [2] -
Chizhova, M.; Dar'in, D.; Krasavin, M. Tetrahedron Lett. 2017, 58, 3470–3473. doi:10.1016/j.tetlet.2017.07.077
Return to citation in text: [1] [2] -
Stanoeva, E.; Haimova, M.; Ognyanov, V. Liebigs Ann. Chem. 1984, 389–394. doi:10.1002/jlac.198419840219
Return to citation in text: [1] [2] [3] -
Kametani, T.; Terasawa, H.; Ihara, M. J. Chem. Soc., Perkin Trans. 1 1976, 2547–2550. doi:10.1039/p19760002547
Return to citation in text: [1] [2] -
Krasavin, M.; Dar’in, D. Tetrahedron Lett. 2016, 57, 1635–1640. doi:10.1016/j.tetlet.2016.03.021
Return to citation in text: [1] -
Valenta, V.; Holubek, J.; Svátek, E.; Dlabač, A.; Bartošová, M.; Protiva, M. Collect. Czech. Chem. Commun. 1983, 48, 1447–1464. doi:10.1135/cccc19831447
Return to citation in text: [1] -
Brossi, A.; Dolan, L. A.; Teitel, S. Org. Synth. 1977, 56, 3. doi:10.15227/orgsyn.056.0003
Return to citation in text: [1] -
Burdzhiev, N. T.; Stanoeva, E. R. Tetrahedron 2006, 62, 8318–8326. doi:10.1016/j.tet.2006.06.054
Return to citation in text: [1] -
Burdzhiev, N.; Stanoeva, E.; Shivachev, B.; Nikolova, R. C. R. Chim. 2014, 17, 420–430. doi:10.1016/j.crci.2013.08.005
Return to citation in text: [1] -
Burdzhiev, N. T.; Stanoeva, E. R. Z. Naturforsch., B: J. Chem. Sci. 2008, 63, 313–320. doi:10.1515/znb-2008-0315
Return to citation in text: [1] -
Dubs, C.; Hamashima, Y.; Sasamoto, N.; Seidel, T. M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. J. Org. Chem. 2008, 73, 5859–5871. doi:10.1021/jo800800y
Return to citation in text: [1] [2] [3] -
Kóbor, J.; Sohár, P.; Fülöp, F. Tetrahedron 1994, 50, 4873–4886. doi:10.1016/s0040-4020(01)85022-5
Return to citation in text: [1] [2] -
Lenz, G. R. J. Org. Chem. 1976, 41, 2201–2207. doi:10.1021/jo00874a029
Return to citation in text: [1] -
Kametani, T.; Takahashi, T.; Honda, T.; Ogasawara, K.; Fukumoto, K. J. Org. Chem. 1974, 39, 447–450. doi:10.1021/jo00918a006
Return to citation in text: [1] -
Yu, C. K.; MacLean, D. B.; Rodrigo, R. G. A.; Manske, R. H. F. Can. J. Chem. 1970, 48, 3673–3678. doi:10.1139/v70-616
Return to citation in text: [1] -
Amat, M.; Santos, M. M. M.; Bassas, O.; Llor, N.; Escolano, C.; Gómez-Esqué, A.; Molins, E.; Allin, S. M.; McKee, V.; Bosch, J. J. Org. Chem. 2007, 72, 5193–5201. doi:10.1021/jo070539g
Return to citation in text: [1] [2] -
Avendaño, C.; Menéndez, J. C. Bicyclic 6-6 Systems with One Bridgehead (Ring Junction) Nitrogen Atom: No Extra Heteroatom. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, 2008; pp 1–75. doi:10.1016/b978-008044992-0.01101-9
Return to citation in text: [1] [2] -
Sugiura, M.; Takao, N.; Iwasa, K.; Sasaki, Y. Chem. Pharm. Bull. 1978, 26, 1901–1907. doi:10.1248/cpb.26.1901
Return to citation in text: [1] -
Fülöp, F.; Bernäth, G.; El-Gharib, M. S.; Kóbor, J.; Sohár, P.; Pelczer, I.; Argay, G.; Kálmán, A. Chem. Ber. 1990, 123, 803–809. doi:10.1002/cber.19901230427
Return to citation in text: [1] -
Zalán, Z.; Hetényi, A.; Lázár, L.; Fülöp, F. Tetrahedron 2005, 61, 5287–5295. doi:10.1016/j.tet.2005.03.067
Return to citation in text: [1] -
Van der Eycken, E.; Deroover, G.; Toppet, S. M.; Hoornaert, G. J. Tetrahedron Lett. 1999, 40, 9147–9150. doi:10.1016/s0040-4039(99)01943-7
Return to citation in text: [1] -
Strandtmann, M. v.; Cohen, M. P.; Shavel, J., Jr. J. Org. Chem. 1966, 31, 797–803. doi:10.1021/jo01341a035
Return to citation in text: [1] -
Sosnovskikh, V. Y.; Usachev, B. I.; Sizov, A. Y.; Vorontsov, I. I.; Shklyaev, Y. V. Org. Lett. 2003, 5, 3123–3126. doi:10.1021/ol0351448
Return to citation in text: [1] -
Tyutin, V. Y.; Chkanikov, N. D.; Shklyaev, V. S.; Shklyaev, Y. V.; Kolomiets, A. F.; Fokin, A. V. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1992, 41, 1474–1476. doi:10.1007/bf00864349
Return to citation in text: [1] -
Sviridov, V. D.; Chkanikov, N. D.; Shklyaev, Y. V.; Kolomiets, A. F.; Fokin, A. V. Chem. Heterocycl. Compd. 1990, 26, 1405. doi:10.1007/bf00473973
Return to citation in text: [1] -
Sviridov, V. D.; Chkanikov, N. D.; Galakhov, M. V.; Shklyaev, Y. V.; Shklyaev, V. S.; Aleksandrov, B. B.; Gavrilov, M. S. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1990, 39, 1268–1272. doi:10.1007/bf00962396
Return to citation in text: [1] -
Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734–736. doi:10.1039/c39760000734
Return to citation in text: [1] -
Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. doi:10.1021/om100106e
Return to citation in text: [1] -
Bruker (2014/5). SADABS-2014/5 Madison, W., USA; Bruker (2013). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1]
| 21. | Stanoeva, E.; Georgieva, A.; Avramova, S.; Burdzhiev, N.; Lázár, L. J. Heterocycl. Chem. 2015, 52, 130–135. doi:10.1002/jhet.1965 |
| 35. | Dubs, C.; Hamashima, Y.; Sasamoto, N.; Seidel, T. M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. J. Org. Chem. 2008, 73, 5859–5871. doi:10.1021/jo800800y |
| 36. | Kóbor, J.; Sohár, P.; Fülöp, F. Tetrahedron 1994, 50, 4873–4886. doi:10.1016/s0040-4020(01)85022-5 |
| 1. | Garrido Montalban, A. Quinolines and Isoquinolines. Heterocycles in Natural Product Synthesis; Wiley-VCH: Weinheim, Germany, 2011; pp 299–339. doi:10.1002/9783527634880.ch9 |
| 6. | Pashev, A. S.; Burdzhiev, N. T.; Stanoeva, E. R. Org. Prep. Proced. Int. 2016, 48, 425–467. doi:10.1080/00304948.2016.1234820 |
| 16. | Ng, P. Y.; Masse, C. E.; Shaw, J. T. Org. Lett. 2006, 8, 3999–4002. doi:10.1021/ol061473z |
| 17. | Di Maso, M. J.; Snyder, K. M.; De Souza Fernandes, F.; Pattawong, O.; Tan, D. Q.; Fettinger, J. C.; Cheong, P. H.-Y.; Shaw, J. T. Chem. – Eur. J. 2016, 22, 4794–4801. doi:10.1002/chem.201504424 |
| 22. | Cushman, M.; Madaj, E. J. J. Org. Chem. 1987, 52, 907–915. doi:10.1021/jo00381a033 |
| 23. | Tang, Y.; Fettinger, J. C.; Shaw, J. T. Org. Lett. 2009, 11, 3802–3805. doi:10.1021/ol901018k |
| 24. | Bakulina, O.; Ivanov, A.; Suslonov, V.; Dar’in, D.; Krasavin, M. Beilstein J. Org. Chem. 2017, 13, 1413–1424. doi:10.3762/bjoc.13.138 |
| 40. | Amat, M.; Santos, M. M. M.; Bassas, O.; Llor, N.; Escolano, C.; Gómez-Esqué, A.; Molins, E.; Allin, S. M.; McKee, V.; Bosch, J. J. Org. Chem. 2007, 72, 5193–5201. doi:10.1021/jo070539g |
| 5. | Zaarur, N.; Gabai, V. L.; Porco, J. A., Jr.; Calderwood, S.; Sherman, M. Y. Cancer Res. 2006, 66, 1783–1791. doi:10.1158/0008-5472.can-05-3692 |
| 24. | Bakulina, O.; Ivanov, A.; Suslonov, V.; Dar’in, D.; Krasavin, M. Beilstein J. Org. Chem. 2017, 13, 1413–1424. doi:10.3762/bjoc.13.138 |
| 47. | Sosnovskikh, V. Y.; Usachev, B. I.; Sizov, A. Y.; Vorontsov, I. I.; Shklyaev, Y. V. Org. Lett. 2003, 5, 3123–3126. doi:10.1021/ol0351448 |
| 48. | Tyutin, V. Y.; Chkanikov, N. D.; Shklyaev, V. S.; Shklyaev, Y. V.; Kolomiets, A. F.; Fokin, A. V. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1992, 41, 1474–1476. doi:10.1007/bf00864349 |
| 49. | Sviridov, V. D.; Chkanikov, N. D.; Shklyaev, Y. V.; Kolomiets, A. F.; Fokin, A. V. Chem. Heterocycl. Compd. 1990, 26, 1405. doi:10.1007/bf00473973 |
| 50. | Sviridov, V. D.; Chkanikov, N. D.; Galakhov, M. V.; Shklyaev, Y. V.; Shklyaev, V. S.; Aleksandrov, B. B.; Gavrilov, M. S. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1990, 39, 1268–1272. doi:10.1007/bf00962396 |
| 3. | Boehringer, M.; Fischer, H.; Hennig, M.; Hunziker, D.; Huwyler, J.; Kuhn, B.; Loeffler, B. M.; Luebbers, T.; Mattei, P.; Narquizian, R.; Sebokova, E.; Sprecher, U.; Wessel, H. P. Bioorg. Med. Chem. Lett. 2010, 20, 1106–1108. doi:10.1016/j.bmcl.2009.12.025 |
| 4. | Mattei, P.; Boehringer, M.; Di Giorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U. Bioorg. Med. Chem. Lett. 2010, 20, 1109–1113. doi:10.1016/j.bmcl.2009.12.024 |
| 13. | Haimova, M. A.; Mollov, N. M.; Ivanova, S. C.; Dimitrova, A. I.; Ognyanov, V. I. Tetrahedron 1977, 33, 331–336. doi:10.1016/0040-4020(77)80114-2 |
| 14. | Cushman, M.; Gentry, J.; Dekow, F. W. J. Org. Chem. 1977, 42, 1111–1116. doi:10.1021/jo00427a001 |
| 15. | Shaw, J. T. Nat. Prod. Rep. 2009, 26, 11–26. doi:10.1039/b814468k |
| 16. | Ng, P. Y.; Masse, C. E.; Shaw, J. T. Org. Lett. 2006, 8, 3999–4002. doi:10.1021/ol061473z |
| 17. | Di Maso, M. J.; Snyder, K. M.; De Souza Fernandes, F.; Pattawong, O.; Tan, D. Q.; Fettinger, J. C.; Cheong, P. H.-Y.; Shaw, J. T. Chem. – Eur. J. 2016, 22, 4794–4801. doi:10.1002/chem.201504424 |
| 18. | Tan, D. Q.; Younai, A.; Pattawong, O.; Fettinger, J. C.; Cheong, P. H.-Y.; Shaw, J. T. Org. Lett. 2013, 15, 5126–5129. doi:10.1021/ol402554n |
| 19. | Pattawong, O.; Tan, D. Q.; Fettinger, J. C.; Shaw, J. T.; Cheong, P. H.-Y. Org. Lett. 2013, 15, 5130–5133. doi:10.1021/ol402561q |
| 36. | Kóbor, J.; Sohár, P.; Fülöp, F. Tetrahedron 1994, 50, 4873–4886. doi:10.1016/s0040-4020(01)85022-5 |
| 41. | Avendaño, C.; Menéndez, J. C. Bicyclic 6-6 Systems with One Bridgehead (Ring Junction) Nitrogen Atom: No Extra Heteroatom. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, 2008; pp 1–75. doi:10.1016/b978-008044992-0.01101-9 |
| 42. | Sugiura, M.; Takao, N.; Iwasa, K.; Sasaki, Y. Chem. Pharm. Bull. 1978, 26, 1901–1907. doi:10.1248/cpb.26.1901 |
| 43. | Fülöp, F.; Bernäth, G.; El-Gharib, M. S.; Kóbor, J.; Sohár, P.; Pelczer, I.; Argay, G.; Kálmán, A. Chem. Ber. 1990, 123, 803–809. doi:10.1002/cber.19901230427 |
| 44. | Zalán, Z.; Hetényi, A.; Lázár, L.; Fülöp, F. Tetrahedron 2005, 61, 5287–5295. doi:10.1016/j.tet.2005.03.067 |
| 45. | Van der Eycken, E.; Deroover, G.; Toppet, S. M.; Hoornaert, G. J. Tetrahedron Lett. 1999, 40, 9147–9150. doi:10.1016/s0040-4039(99)01943-7 |
| 46. | Strandtmann, M. v.; Cohen, M. P.; Shavel, J., Jr. J. Org. Chem. 1966, 31, 797–803. doi:10.1021/jo01341a035 |
| 2. | Dwek, R. A.; Butters, T. D.; Platt, F. M.; Zitzmann, N. Nat. Rev. Drug Discovery 2002, 1, 65–75. doi:10.1038/nrd708 |
| 13. | Haimova, M. A.; Mollov, N. M.; Ivanova, S. C.; Dimitrova, A. I.; Ognyanov, V. I. Tetrahedron 1977, 33, 331–336. doi:10.1016/0040-4020(77)80114-2 |
| 14. | Cushman, M.; Gentry, J.; Dekow, F. W. J. Org. Chem. 1977, 42, 1111–1116. doi:10.1021/jo00427a001 |
| 20. | Georgieva, A.; Stanoeva, E.; Spassov, S.; Macicek, J.; Angelova, O.; Haimova, M.; De Kimpe, N. Tetrahedron 1991, 47, 3375–3388. doi:10.1016/s0040-4020(01)86402-4 |
| 21. | Stanoeva, E.; Georgieva, A.; Avramova, S.; Burdzhiev, N.; Lázár, L. J. Heterocycl. Chem. 2015, 52, 130–135. doi:10.1002/jhet.1965 |
| 35. | Dubs, C.; Hamashima, Y.; Sasamoto, N.; Seidel, T. M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. J. Org. Chem. 2008, 73, 5859–5871. doi:10.1021/jo800800y |
| 11. | Shetty, B. V.; McFadden, A.; Hofer, P. Analgesic 4-carboxy-pyrrolidin-2-one compound. U.S. Patent US4,476,311, Oct 9, 1984. |
| 12. | Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014 |
| 40. | Amat, M.; Santos, M. M. M.; Bassas, O.; Llor, N.; Escolano, C.; Gómez-Esqué, A.; Molins, E.; Allin, S. M.; McKee, V.; Bosch, J. J. Org. Chem. 2007, 72, 5193–5201. doi:10.1021/jo070539g |
| 10. | Cushman, M.; Castagnoli, N. J. Org. Chem. 1973, 38, 440–448. doi:10.1021/jo00943a007 |
| 13. | Haimova, M. A.; Mollov, N. M.; Ivanova, S. C.; Dimitrova, A. I.; Ognyanov, V. I. Tetrahedron 1977, 33, 331–336. doi:10.1016/0040-4020(77)80114-2 |
| 14. | Cushman, M.; Gentry, J.; Dekow, F. W. J. Org. Chem. 1977, 42, 1111–1116. doi:10.1021/jo00427a001 |
| 41. | Avendaño, C.; Menéndez, J. C. Bicyclic 6-6 Systems with One Bridgehead (Ring Junction) Nitrogen Atom: No Extra Heteroatom. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, 2008; pp 1–75. doi:10.1016/b978-008044992-0.01101-9 |
| 14. | Cushman, M.; Gentry, J.; Dekow, F. W. J. Org. Chem. 1977, 42, 1111–1116. doi:10.1021/jo00427a001 |
| 37. | Lenz, G. R. J. Org. Chem. 1976, 41, 2201–2207. doi:10.1021/jo00874a029 |
| 38. | Kametani, T.; Takahashi, T.; Honda, T.; Ogasawara, K.; Fukumoto, K. J. Org. Chem. 1974, 39, 447–450. doi:10.1021/jo00918a006 |
| 39. | Yu, C. K.; MacLean, D. B.; Rodrigo, R. G. A.; Manske, R. H. F. Can. J. Chem. 1970, 48, 3673–3678. doi:10.1139/v70-616 |
| 7. | Matveeva, M. D.; Purgatorio, R.; Voskressensky, L. G.; Altomare, C. D. Future Med. Chem. 2019, 11, 2735–2755. doi:10.4155/fmc-2019-0136 |
| 8. | Pässler, U.; Knölker, H.-J. The Pyrrolo[2,1-a]isoquinoline Alkaloids. Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, Netherlands, 2011; Vol. 70, pp 79–151. doi:10.1016/b978-0-12-391426-2.00002-5 |
| 11. | Shetty, B. V.; McFadden, A.; Hofer, P. Analgesic 4-carboxy-pyrrolidin-2-one compound. U.S. Patent US4,476,311, Oct 9, 1984. |
| 35. | Dubs, C.; Hamashima, Y.; Sasamoto, N.; Seidel, T. M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. J. Org. Chem. 2008, 73, 5859–5871. doi:10.1021/jo800800y |
| 13. | Haimova, M. A.; Mollov, N. M.; Ivanova, S. C.; Dimitrova, A. I.; Ognyanov, V. I. Tetrahedron 1977, 33, 331–336. doi:10.1016/0040-4020(77)80114-2 |
| 21. | Stanoeva, E.; Georgieva, A.; Avramova, S.; Burdzhiev, N.; Lázár, L. J. Heterocycl. Chem. 2015, 52, 130–135. doi:10.1002/jhet.1965 |
| 24. | Bakulina, O.; Ivanov, A.; Suslonov, V.; Dar’in, D.; Krasavin, M. Beilstein J. Org. Chem. 2017, 13, 1413–1424. doi:10.3762/bjoc.13.138 |
| 25. | González-López, M.; Shaw, J. T. Chem. Rev. 2009, 109, 164–189. doi:10.1021/cr8002714 |
| 24. | Bakulina, O.; Ivanov, A.; Suslonov, V.; Dar’in, D.; Krasavin, M. Beilstein J. Org. Chem. 2017, 13, 1413–1424. doi:10.3762/bjoc.13.138 |
| 51. | Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734–736. doi:10.1039/c39760000734 |
| 16. | Ng, P. Y.; Masse, C. E.; Shaw, J. T. Org. Lett. 2006, 8, 3999–4002. doi:10.1021/ol061473z |
| 23. | Tang, Y.; Fettinger, J. C.; Shaw, J. T. Org. Lett. 2009, 11, 3802–3805. doi:10.1021/ol901018k |
| 52. | Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. doi:10.1021/om100106e |
| 53. | Bruker (2014/5). SADABS-2014/5 Madison, W., USA; Bruker (2013). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA. |
| 30. | Valenta, V.; Holubek, J.; Svátek, E.; Dlabač, A.; Bartošová, M.; Protiva, M. Collect. Czech. Chem. Commun. 1983, 48, 1447–1464. doi:10.1135/cccc19831447 |
| 31. | Brossi, A.; Dolan, L. A.; Teitel, S. Org. Synth. 1977, 56, 3. doi:10.15227/orgsyn.056.0003 |
| 32. | Burdzhiev, N. T.; Stanoeva, E. R. Tetrahedron 2006, 62, 8318–8326. doi:10.1016/j.tet.2006.06.054 |
| 33. | Burdzhiev, N.; Stanoeva, E.; Shivachev, B.; Nikolova, R. C. R. Chim. 2014, 17, 420–430. doi:10.1016/j.crci.2013.08.005 |
| 34. | Burdzhiev, N. T.; Stanoeva, E. R. Z. Naturforsch., B: J. Chem. Sci. 2008, 63, 313–320. doi:10.1515/znb-2008-0315 |
| 12. | Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014 |
| 27. | Stanoeva, E.; Haimova, M.; Ognyanov, V. Liebigs Ann. Chem. 1984, 389–394. doi:10.1002/jlac.198419840219 |
| 26. | Chizhova, M.; Dar'in, D.; Krasavin, M. Tetrahedron Lett. 2017, 58, 3470–3473. doi:10.1016/j.tetlet.2017.07.077 |
| 27. | Stanoeva, E.; Haimova, M.; Ognyanov, V. Liebigs Ann. Chem. 1984, 389–394. doi:10.1002/jlac.198419840219 |
| 12. | Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014 |
| 12. | Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014 |
| 25. | González-López, M.; Shaw, J. T. Chem. Rev. 2009, 109, 164–189. doi:10.1021/cr8002714 |
| 26. | Chizhova, M.; Dar'in, D.; Krasavin, M. Tetrahedron Lett. 2017, 58, 3470–3473. doi:10.1016/j.tetlet.2017.07.077 |
| 27. | Stanoeva, E.; Haimova, M.; Ognyanov, V. Liebigs Ann. Chem. 1984, 389–394. doi:10.1002/jlac.198419840219 |
| 28. | Kametani, T.; Terasawa, H.; Ihara, M. J. Chem. Soc., Perkin Trans. 1 1976, 2547–2550. doi:10.1039/p19760002547 |
| 29. | Krasavin, M.; Dar’in, D. Tetrahedron Lett. 2016, 57, 1635–1640. doi:10.1016/j.tetlet.2016.03.021 |
| 54. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 28. | Kametani, T.; Terasawa, H.; Ihara, M. J. Chem. Soc., Perkin Trans. 1 1976, 2547–2550. doi:10.1039/p19760002547 |
| 55. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
© 2020 Pashev et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)