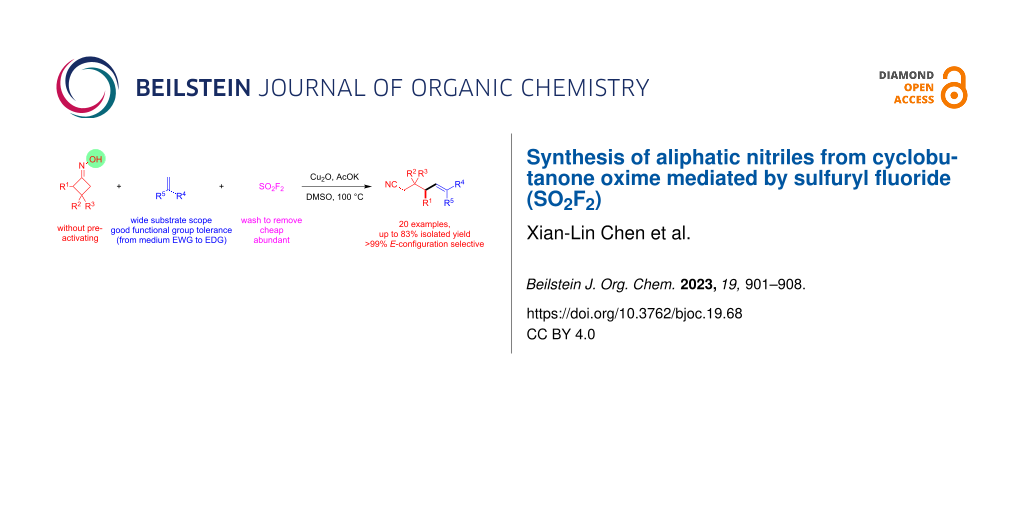
Abstract
A SO2F2-mediated ring-opening cross-coupling of cyclobutanone oxime derivatives with alkenes was developed for the construction of a range of δ-olefin-containing aliphatic nitriles with (E)-configuration selectivity. This new method features wide substrate scope, mild conditions, and direct N–O activation.

Graphical Abstract
Introduction
As an important functional group in organic molecules, the nitrile group is commonly present in functional materials [1,2], nanoscale drug carriers [3-5], biologically valuable molecules and drugs (Scheme 1) [6,7]. There are over 70 nitrile-containing drugs approved by the FDA for various indications and more than 140 additional nitrile-containing leads in clinical investigation [8]. Looking into changing the physicochemical properties, in the field of drug discovery, it is important to explore solutions to introduce nitrile groups into a molecule for enhancing the interaction between the drug candidate and the target protein, to further improve the efficacy of the potential drug [9]. The nitrile group can also function as a metabolic blocking site to inhibit the oxidative metabolism of molecules to improve metabolic stability in vivo [10]. Consequently, the development of novel synthetic methods and strategies toward nitrile group construction continues to be a focus for synthetic chemists.
![[1860-5397-19-68-i1]](/bjoc/content/inline/1860-5397-19-68-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative nitrile-containing functional materials, drug carriers, and medicines.
Scheme 1: Representative nitrile-containing functional materials, drug carriers, and medicines.
The cross-coupling reactions of C–C bonds catalyzed by transition-metal complexes play a crucial role in modern organic synthesis, as they make it feasible to synthesize complex structures from available components [11-13]. Indeed, the formation of C(sp2)–C(sp3) bonds by cross-coupling has developed rapidly in recent years [14-16], but it still remains less advanced than the synthesis of C(sp2)–C(sp2) bonds [17,18]. This is attributed to the electron-richness of the C(sp3) carbon, which leads to side reactions of the alkyl intermediates [14,19,20]. Besides, most of the C(sp2)–C(sp3) reactions employ organic halides or organometallic reagents [21-23], which are not environmentally friendly.
Recently, based on the activation effect of O-acyloximes on N–O bonds [24-29], a synthesis method for δ-olefin-containing aliphatic nitriles by the radical C–C bond cleavage of cycloketone oxime ester derivatives was developed by Shi’s group (Scheme 2a) [30], which emerged as an efficient strategy to construct C(sp2)–C(sp3) bonds [31-33]. Later, Xiao [34], Liu [35], and Yang [36] achieved similar transformations through visible-light photocatalysis. In addition, Guo [37,38] improved the protocol by using low-cost nickel and iron catalysts. However, most of these advancements mainly relied on the excellent redox potential manipulation of cyclic oxime esters and adopted the pre-acylation activation strategies [39-41]. Up to now, only one report employed an oxime for the generation of iminyl radicals to obtain the similar products, in which, substrates were limited to the electron-rich alkenes (Scheme 2b) [42].
![[1860-5397-19-68-i2]](/bjoc/content/inline/1860-5397-19-68-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Activating protocol of cyclobutanone oximes.
Scheme 2: Activating protocol of cyclobutanone oximes.
On the other hand, sulfuryl fluoride (SO2F2) [43], a kind of inexpensive (about 1 $/kg), abundant, and relatively inert electrophile and one of the major sulfur fluoride exchange (SuFEx) click chemistry reagents [44,45], has been successfully applied as an electrophile to react with hydroxy groups to generate fluorosulfonate esters, being activated intermediates for a variety of transformations [46-58]. Lately, we discovered the SO2F2-mediated transformation of primary alcohols to nitriles, involving an aldoxime sulfonyl ester intermediate (Scheme 2c) [59]. Drawing inspiration from these excellent works, we contemplated that the N–O bond of cyclobutanone oxime derivatives could be activated by SO2F2 in situ to enable cleavage of the C–C bond, which could achieve this transformation without going through inefficient pre-introduction of electrophores. Herein, we describe how this concept has been translated into experimental reality, developing a new SO2F2-mediated C–C single bond cleavage method for constructing δ-olefin-containing aliphatic nitriles.
Results and Discussion
We started our investigation by selecting cyclobutanone oxime (1a) and 1,1-diphenylethylene (2a) as model starting materials to testify the feasibility of this proposed transformation in the presence of N,N-diisopropylethylamine (DIPEA) and Cu(OTf)2 in dioxane/PhCF3 (1:1) under an SO2F2 atmosphere at 100 °C. The desired product 6,6-diphenylhex-5-enenitrile (3aa) was obtained in 24% yield (Table 1, entry 1) and according to the control experiment, SO2F2 is essential for the reaction to proceed (Table 1, entry 2). Encouraged by the preliminary result, we then screened a large variety of conditions as shown in Table 1 in order to improve the efficiency of the transformation. The investigation of the solvent effect revealed that in 1,4-dioxane the transformations performed the best (Table 1, entries 3–5). A series of copper catalysts such as CuI, CuCN, and Cu2O was screened, in which some showed good catalytic activity (Table 1, entries 6–9), and Cu2O was identified as the most effective catalyst for the desired transformation. Accordingly, the catalyst loading of Cu2O was studied next and increasing the loading of Cu2O to 1.0 equivalent, gave the desired product 3aa in a good yield of 72% (Table 1, entry 10). Furthermore, the examination of the effect of base revealed CH3COOK being the most suitable choice (Table 1, entry 11). Either increasing the temperature to 120 °C or decreasing to 80 °C resulted in an obviously decreased yield (Table 1, entries 13 and 14), which could probably be attributed to the decomposition of the highly active sulfonyl ester intermediate. The reaction time was also screened and the yield did not change within the accuracy errors when the time was extended (Table 1, entry 15) and among the screened reaction times, 12 hours were chosen as the optimal conditions (see Supporting Information File 1 for more details).
Table 1: Screening the optimized reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-68-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | [Cu] cat. | Base | Solvent | T (°C) | Yield (%)b |
| 1c | Cu(OTf)2 (10 mol %) | DIPEA | dioxane/PhCF3 | 100 | 24 |
| 2c,d | Cu(OTf)2 (10 mol %) | DIPEA | dioxane/PhCF3 | 100 | N.D. |
| 3c | Cu(OTf)2 (10 mol %) | DIPEA | PhCF3 | 100 | N.D. |
| 4c | Cu(OTf)2 (10 mol %) | DIPEA | 1,4-dioxane | 100 | 47 |
| 5c | Cu(OTf)2 (10 mol %) | DIPEA | CH2Cl2 | 100 | 39 |
| 6c | CuI (10 mol %) | DIPEA | 1,4-dioxane | 100 | 41 |
| 7c | CuCN (10 mol %) | DIPEA | 1,4-dioxane | 100 | 40 |
| 8c | Cu2O (10 mol %) | DIPEA | 1,4-dioxane | 100 | 55 |
| 9c | / | DIPEA | 1,4-dioxane | 100 | N.D. |
| 10c | Cu2O (100 mol %) | DIPEA | 1,4-dioxane | 100 | 72 |
| 11c | Cu2O (100 mol %) | CH3COOK | 1,4-dioxane | 100 | 75 |
| 12 | Cu2O (100 mol %) | CH3COOK | 1,4-dioxane | 100 | 83 |
| 13 | Cu2O (100 mol %) | CH3COOK | 1,4-dioxane | 80 | 54 |
| 14 | Cu2O (100 mol %) | CH3COOK | 1,4-dioxane | 120 | 64 |
| 15e | Cu2O (100 mol %) | CH3COOK | 1,4-dioxane | 100 | 79 |
aReaction conditions: A mixture of cyclobutanone oxime (1a, 1.5 mmol, 3.0 equiv), 1,1-diphenylethylene (2a, 0.5 mmol, 1.0 equiv), copper catalyst and base (5.0 mmol, 10.0 equiv) in extra dry solvent (0.1 M) was stirred at the corresponding temperature under an SO2F2 atmosphere (balloon) for 12 h. bThe yield was determined by HPLC using pure 3aa as the external standard (tR = 5.017 min, λmax = 250.0 nm, water/methanol 20:80 (v/v)). c6.0 equiv of base were used. dUnder Ar atmosphere (balloon) instead of SO2F2. eThe reaction lasted 16 h.
With the optimized reaction conditions in hand, a range of other substrates possessing representative functional groups was employed subsequently to evaluate the reaction scope and limitations (Scheme 3). Under the optimized conditions, alkenes 2b–e with varying steric effects underwent smooth reaction, yielding the corresponding products 3ab–ae in moderate to good yields (56–68%). Notably, the efficiency of this transformation was greatly impacted by the electronic effect on the aromatic rings of the olefins. Alkenes with electron-donating groups on their aromatic rings showed higher yields of the corresponding products as compared to those with electron-withdrawing groups (3af–ar). Furthermore, the desired products were not even obtained when the starting materials were connected to extra strong electron-withdrawing groups (such as a nitro group) on their aromatic rings. In addition, a series of cyclobutanone oxime derivatives were also smoothly transformed into the corresponding nitriles 3ba–da in excellent yields.
![[1860-5397-19-68-i3]](/bjoc/content/inline/1860-5397-19-68-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of δ-olefin-containing aliphatic nitriles. Reaction conditions: A mixture of cyclobutanone oxime derivative 1 (3.0 mmol, 3.0 equiv), alkene 2 (1.0 mmol, 1.0 equiv), Cu2O (1.0 mmol, 1.0 equiv) and potassium acetate (10.0 mmol, 10.0 equiv) in extra dry dioxane or DMSO (0.1 M) was stirred at 100 °C under an SO2F2 atmosphere (balloon) for 12 h; yields refer to isolated compounds.
Scheme 3: Substrate scope of δ-olefin-containing aliphatic nitriles. Reaction conditions: A mixture of cyclob...
Interestingly, when the loading of CH3COOK was reduced to 2 equivalents, we obtained a mixture of unsaturated nitrile 3aa and saturated nitrile 4 after column chromatography (Scheme 4). We speculated that the reduction of the base equivalent may induce the ionization of 1a and facilitate the ultimate addition process. The selectivity of bases for different processes may attract significant attention for further applications.
![[1860-5397-19-68-i4]](/bjoc/content/inline/1860-5397-19-68-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Competition between two reactions caused by the reduction of base equivalent.
Scheme 4: Competition between two reactions caused by the reduction of base equivalent.
In order to understand the mechanism of the aforementioned transformation, some experimental investigations were performed as described in Scheme 5. Under the promotion of the base, cyclobutanone oxime preliminarily reacts with SO2F2, generating the activated precursor fluorosulfonate, which further reacts with the alkene 2a in the presence of the copper catalyst under Ar atmosphere for 9 h (Scheme 5a). The corresponding product was successfully obtained in 45% yield, which indirectly proved the existence of an oxime sulfonyl ester intermediate (fluorosulfonate). As shown in Scheme 5b, in the presence of one equivalent of TEMPO, a commonly used radical scavenger, the yield of 3aa significantly decreased, in addition, the reaction was completely inhibited when the amount of added TEMPO was increased to 2 equivalents.
![[1860-5397-19-68-i5]](/bjoc/content/inline/1860-5397-19-68-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based upon the preliminary results and previous reports of this class of transformation [26,30,36,37,42,60,61], a plausible mechanism for the base-promoted, SO2F2-mediated ring-opening cross-coupling of cyclobutanone oxime derivatives with alkenes was proposed (Scheme 6). Initially, cyclobutanone oxime reacts with SO2F2, generating an oxime sulfonyl ester intermediate (fluorosulfonate) I promoted by the base. Subsequently, the intermediate fluorosulfonate I undergoes single-electron reduction by [Cun] in situ to afford the iminyl radical intermediate II. In the following step, the ring-strain of cyclobutanone is released under the promotion of the imine radical, giving the C-centered radical III which is subsequently captured by the alkene. Meanwhile, the radical IV transfers an electron to [Cun+1] regenerating the [Cun] catalyst and intermediate V. The critical β-H elimination step occurs smoothly in the presence of excessive base to generate the final nitrile product. Due to the high reactivity of the intermediate fluorosulfonate I, the attempt of isolation or detecting the in situ generated intermediate I was not accomplished either by NMR analysis or chromatography.
![[1860-5397-19-68-i6]](/bjoc/content/inline/1860-5397-19-68-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: A proposed plausible mechanism.
Scheme 6: A proposed plausible mechanism.
Conclusion
In conclusion, we have developed an SO2F2-mediated ring-opening cross-coupling reaction of cyclobutanone oxime derivatives with alkenes for the synthesis of a class of novel elongated nitriles. The newly constructed δ-olefin-containing aliphatic nitriles possess E-configuration at the double bond. This transformation could be easily activated by SO2F2 in situ without the need of pre-introduction of electrophores.
Supporting Information
| Supporting Information File 1: Experimental information. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
An, B.-K.; Gierschner, J.; Park, S. Y. Acc. Chem. Res. 2012, 45, 544–554. doi:10.1021/ar2001952
Return to citation in text: [1] -
Gierschner, J.; Park, S. Y. J. Mater. Chem. C 2013, 1, 5818–5832. doi:10.1039/c3tc31062k
Return to citation in text: [1] -
Couvreur, P. J. Controlled Release 2021, 334, 318–326. doi:10.1016/j.jconrel.2021.04.028
Return to citation in text: [1] -
Kim, K. Y.; Jin, H.; Park, J.; Jung, S. H.; Lee, J. H.; Park, H.; Kim, S. K.; Bae, J.; Jung, J. H. Nano Res. 2018, 11, 1082–1098. doi:10.1007/s12274-017-1728-7
Return to citation in text: [1] -
Zhang, W.; Huo, F.; Yin, C. J. Mater. Chem. B 2018, 6, 6919–6929. doi:10.1039/c8tb02205d
Return to citation in text: [1] -
Barradas, M. A.; Jagroop, A.; O'Donoghue, S.; Jeremy, J. Y.; Mikhailidis, D. P. Thromb. Res. 1993, 71, 227–236. doi:10.1016/0049-3848(93)90097-8
Return to citation in text: [1] -
Noble, S.; Benfield, P. CNS Drugs 1997, 8, 410–431. doi:10.2165/00023210-199708050-00009
Return to citation in text: [1] -
Wishart, D. S.; Feunang, Y. D.; Guo, A. C.; Lo, E. J.; Marcu, A.; Grant, J. R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; Assempour, N.; Iynkkaran, I.; Liu, Y.; Maciejewski, A.; Gale, N.; Wilson, A.; Chin, L.; Cummings, R.; Le, D.; Pon, A.; Knox, C.; Wilson, M. Nucleic Acids Res. 2018, 46, D1074–D1082. doi:10.1093/nar/gkx1037
Return to citation in text: [1] -
Wang, Y.; Du, Y.; Huang, N. Future Med. Chem. 2018, 10, 2713–2728. doi:10.4155/fmc-2018-0252
Return to citation in text: [1] -
Fleming, F. F.; Yao, L.; Ravikumar, P. C.; Funk, L.; Shook, B. C. J. Med. Chem. 2010, 53, 7902–7917. doi:10.1021/jm100762r
Return to citation in text: [1] -
Jun, C.-H. Chem. Soc. Rev. 2004, 33, 610–618. doi:10.1039/b308864m
Return to citation in text: [1] -
Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613–8661. doi:10.1021/cr400628s
Return to citation in text: [1] -
Dermenci, A.; Coe, J. W.; Dong, G. Org. Chem. Front. 2014, 1, 567–581. doi:10.1039/c4qo00053f
Return to citation in text: [1] -
Frisch, A. C.; Beller, M. Angew. Chem., Int. Ed. 2005, 44, 674–688. doi:10.1002/anie.200461432
Return to citation in text: [1] [2] -
Rudolph, A.; Lautens, M. Angew. Chem., Int. Ed. 2009, 48, 2656–2670. doi:10.1002/anie.200803611
Return to citation in text: [1] -
Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417–1492. doi:10.1021/cr100327p
Return to citation in text: [1] -
Huihui, K. M. M.; Caputo, J. A.; Melchor, Z.; Olivares, A. M.; Spiewak, A. M.; Johnson, K. A.; DiBenedetto, T. A.; Kim, S.; Ackerman, L. K. G.; Weix, D. J. J. Am. Chem. Soc. 2016, 138, 5016–5019. doi:10.1021/jacs.6b01533
Return to citation in text: [1] -
Miyaura, N.; Yanagi, T.; Suzuki, A. Synth. Commun. 1981, 11, 513–519. doi:10.1080/00397918108063618
Return to citation in text: [1] -
Terao, J.; Kambe, N. Bull. Chem. Soc. Jpn. 2006, 79, 663–672. doi:10.1246/bcsj.79.663
Return to citation in text: [1] -
Sherry, B. D.; Fürstner, A. Acc. Chem. Res. 2008, 41, 1500–1511. doi:10.1021/ar800039x
Return to citation in text: [1] -
Bloome, K. S.; McMahen, R. L.; Alexanian, E. J. J. Am. Chem. Soc. 2011, 133, 20146–20148. doi:10.1021/ja2091883
Return to citation in text: [1] -
Pearson, R. G.; Figdore, P. E. J. Am. Chem. Soc. 1980, 102, 1541–1547. doi:10.1021/ja00525a013
Return to citation in text: [1] -
Alisha, M.; Philip, R. M.; Anilkumar, G. J. Organomet. Chem. 2022, 959, 122207. doi:10.1016/j.jorganchem.2021.122207
Return to citation in text: [1] -
Nishimura, T.; Uemura, S. J. Am. Chem. Soc. 2000, 122, 12049–12050. doi:10.1021/ja005558l
Return to citation in text: [1] -
Nishimura, T.; Yoshinaka, T.; Nishiguchi, Y.; Maeda, Y.; Uemura, S. Org. Lett. 2005, 7, 2425–2427. doi:10.1021/ol0507120
Return to citation in text: [1] -
Yang, H.-B.; Selander, N. Chem. – Eur. J. 2017, 23, 1779–1783. doi:10.1002/chem.201605636
Return to citation in text: [1] [2] -
Faulkner, A.; Race, N. J.; Scott, J. S.; Bower, J. F. Chem. Sci. 2014, 5, 2416–2421. doi:10.1039/c4sc00652f
Return to citation in text: [1] -
Lin, X.; Stien, D.; Weinreb, S. M. Org. Lett. 1999, 1, 637–640. doi:10.1021/ol990720e
Return to citation in text: [1] -
Krylov, I. B.; Segida, O. O.; Budnikov, A. S.; Terent'ev, A. O. Adv. Synth. Catal. 2021, 363, 2502–2528. doi:10.1002/adsc.202100058
Return to citation in text: [1] -
Zhao, B.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 12727–12731. doi:10.1002/anie.201707181
Return to citation in text: [1] [2] -
Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron Lett. 1991, 32, 4299–4302. doi:10.1016/s0040-4039(00)92153-1
Return to citation in text: [1] -
Yang, H.-B.; Pathipati, S. R.; Selander, N. ACS Catal. 2017, 7, 8441–8445. doi:10.1021/acscatal.7b03432
Return to citation in text: [1] -
Boivin, J.; Fouquet, E.; Zard, S. Z. J. Am. Chem. Soc. 1991, 113, 1055–1057. doi:10.1021/ja00003a057
Return to citation in text: [1] -
Yu, X.-Y.; Chen, J.-R.; Wang, P.-Z.; Yang, M.-N.; Liang, D.; Xiao, W.-J. Angew. Chem., Int. Ed. 2018, 57, 738–743. doi:10.1002/anie.201710618
Return to citation in text: [1] -
Tu, J.-L.; Tang, W.; Xu, W.; Liu, F. J. Org. Chem. 2021, 86, 2929–2940. doi:10.1021/acs.joc.0c02834
Return to citation in text: [1] -
Gao, J.; Ye, Z.-P.; Liu, Y.-F.; He, X.-C.; Guan, J.-P.; Liu, F.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2022, 24, 4640–4644. doi:10.1021/acs.orglett.2c01750
Return to citation in text: [1] [2] -
Zhao, J.-F.; Duan, X.-H.; Gu, Y.-R.; Gao, P.; Guo, L.-N. Org. Lett. 2018, 20, 4614–4617. doi:10.1021/acs.orglett.8b01901
Return to citation in text: [1] [2] -
Gu, Y.-R.; Duan, X.-H.; Yang, L.; Guo, L.-N. Org. Lett. 2017, 19, 5908–5911. doi:10.1021/acs.orglett.7b02902
Return to citation in text: [1] -
Xiao, T.; Zhou, L.; Huang, H.; Anand, D. Synthesis 2020, 52, 1585–1601. doi:10.1055/s-0039-1690844
Return to citation in text: [1] -
Liu, L.; Duan, X.-H.; Guo, L.-N. Synthesis 2021, 53, 4375–4388. doi:10.1055/a-1545-6874
Return to citation in text: [1] -
Lu, X.-Y.; Xia, Z.-J.; Gao, A.; Liu, Q.-L.; Jiang, R.-C.; Liu, C.-C. J. Org. Chem. 2021, 86, 8829–8842. doi:10.1021/acs.joc.1c00726
Return to citation in text: [1] -
Xia, P.-J.; Ye, Z.-P.; Hu, Y.-Z.; Song, D.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2019, 21, 2658–2662. doi:10.1021/acs.orglett.9b00651
Return to citation in text: [1] [2] -
Sulbaek Andersen, M. P.; Blake, D. R.; Rowland, F. S.; Hurley, M. D.; Wallington, T. J. Environ. Sci. Technol. 2009, 43, 1067–1070. doi:10.1021/es802439f
Return to citation in text: [1] -
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Return to citation in text: [1] -
Qin, H.-L.; Zheng, Q.; Bare, G. A. L.; Wu, P.; Sharpless, K. B. Angew. Chem., Int. Ed. 2016, 55, 14155–14158. doi:10.1002/anie.201608807
Return to citation in text: [1] -
Gao, B.; Zhang, L.; Zheng, Q.; Zhou, F.; Klivansky, L. M.; Lu, J.; Liu, Y.; Dong, J.; Wu, P.; Sharpless, K. B. Nat. Chem. 2017, 9, 1083–1088. doi:10.1038/nchem.2796
Return to citation in text: [1] -
Lekkala, R.; Lekkala, R.; Moku, B.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2019, 6, 3490–3516. doi:10.1039/c9qo00747d
Return to citation in text: [1] -
Liu, J.; Wang, S.-M.; Qin, H.-L. Tetrahedron 2020, 76, 131724. doi:10.1016/j.tet.2020.131724
Return to citation in text: [1] -
Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a
Return to citation in text: [1] -
Liu, J.; Wang, S.-M.; Alharbi, N. S.; Qin, H.-L. Beilstein J. Org. Chem. 2019, 15, 1907–1912. doi:10.3762/bjoc.15.186
Return to citation in text: [1] -
Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Alharbi, N. S.; Qin, H.-L. Tetrahedron 2019, 75, 4648–4656. doi:10.1016/j.tet.2019.07.007
Return to citation in text: [1] -
Fang, W.-Y.; Zha, G.-F.; Zhao, C.; Qin, H.-L. Chem. Commun. 2019, 55, 6273–6276. doi:10.1039/c9cc02659b
Return to citation in text: [1] -
Wang, S.-M.; Alharbi, N. S.; Qin, H.-L. Synthesis 2019, 51, 3901–3907. doi:10.1055/s-0039-1690017
Return to citation in text: [1] -
Fang, W.-Y.; Qin, H.-L. J. Org. Chem. 2019, 84, 5803–5812. doi:10.1021/acs.joc.8b03164
Return to citation in text: [1] -
Epifanov, M.; Foth, P. J.; Gu, F.; Barrillon, C.; Kanani, S. S.; Higman, C. S.; Hein, J. E.; Sammis, G. M. J. Am. Chem. Soc. 2018, 140, 16464–16468. doi:10.1021/jacs.8b11309
Return to citation in text: [1] -
Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891
Return to citation in text: [1] -
Hanley, P. S.; Clark, T. P.; Krasovskiy, A. L.; Ober, M. S.; O’Brien, J. P.; Staton, T. S. ACS Catal. 2016, 6, 3515–3519. doi:10.1021/acscatal.6b00865
Return to citation in text: [1] -
Zhang, E.; Tang, J.; Li, S.; Wu, P.; Moses, J. E.; Sharpless, K. B. Chem. – Eur. J. 2016, 22, 5692–5697. doi:10.1002/chem.201600167
Return to citation in text: [1] -
Jiang, Y.; Sun, B.; Fang, W.-Y.; Qin, H.-L. Eur. J. Org. Chem. 2019, 3190–3194. doi:10.1002/ejoc.201900478
Return to citation in text: [1] -
Li, L.; Chen, H.; Mei, M.; Zhou, L. Chem. Commun. 2017, 53, 11544–11547. doi:10.1039/c7cc07347j
Return to citation in text: [1] -
Liu, X.-F.; Zhang, K.; Wang, L.-L.; Wang, H.; Huang, J.; Zhang, X.-T.; Lu, X.-B.; Zhang, W.-Z. J. Org. Chem. 2023, 88, 5212–5219. doi:10.1021/acs.joc.2c01816
Return to citation in text: [1]
| 1. | An, B.-K.; Gierschner, J.; Park, S. Y. Acc. Chem. Res. 2012, 45, 544–554. doi:10.1021/ar2001952 |
| 2. | Gierschner, J.; Park, S. Y. J. Mater. Chem. C 2013, 1, 5818–5832. doi:10.1039/c3tc31062k |
| 9. | Wang, Y.; Du, Y.; Huang, N. Future Med. Chem. 2018, 10, 2713–2728. doi:10.4155/fmc-2018-0252 |
| 34. | Yu, X.-Y.; Chen, J.-R.; Wang, P.-Z.; Yang, M.-N.; Liang, D.; Xiao, W.-J. Angew. Chem., Int. Ed. 2018, 57, 738–743. doi:10.1002/anie.201710618 |
| 8. | Wishart, D. S.; Feunang, Y. D.; Guo, A. C.; Lo, E. J.; Marcu, A.; Grant, J. R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; Assempour, N.; Iynkkaran, I.; Liu, Y.; Maciejewski, A.; Gale, N.; Wilson, A.; Chin, L.; Cummings, R.; Le, D.; Pon, A.; Knox, C.; Wilson, M. Nucleic Acids Res. 2018, 46, D1074–D1082. doi:10.1093/nar/gkx1037 |
| 35. | Tu, J.-L.; Tang, W.; Xu, W.; Liu, F. J. Org. Chem. 2021, 86, 2929–2940. doi:10.1021/acs.joc.0c02834 |
| 6. | Barradas, M. A.; Jagroop, A.; O'Donoghue, S.; Jeremy, J. Y.; Mikhailidis, D. P. Thromb. Res. 1993, 71, 227–236. doi:10.1016/0049-3848(93)90097-8 |
| 7. | Noble, S.; Benfield, P. CNS Drugs 1997, 8, 410–431. doi:10.2165/00023210-199708050-00009 |
| 30. | Zhao, B.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 12727–12731. doi:10.1002/anie.201707181 |
| 3. | Couvreur, P. J. Controlled Release 2021, 334, 318–326. doi:10.1016/j.jconrel.2021.04.028 |
| 4. | Kim, K. Y.; Jin, H.; Park, J.; Jung, S. H.; Lee, J. H.; Park, H.; Kim, S. K.; Bae, J.; Jung, J. H. Nano Res. 2018, 11, 1082–1098. doi:10.1007/s12274-017-1728-7 |
| 5. | Zhang, W.; Huo, F.; Yin, C. J. Mater. Chem. B 2018, 6, 6919–6929. doi:10.1039/c8tb02205d |
| 31. | Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron Lett. 1991, 32, 4299–4302. doi:10.1016/s0040-4039(00)92153-1 |
| 32. | Yang, H.-B.; Pathipati, S. R.; Selander, N. ACS Catal. 2017, 7, 8441–8445. doi:10.1021/acscatal.7b03432 |
| 33. | Boivin, J.; Fouquet, E.; Zard, S. Z. J. Am. Chem. Soc. 1991, 113, 1055–1057. doi:10.1021/ja00003a057 |
| 17. | Huihui, K. M. M.; Caputo, J. A.; Melchor, Z.; Olivares, A. M.; Spiewak, A. M.; Johnson, K. A.; DiBenedetto, T. A.; Kim, S.; Ackerman, L. K. G.; Weix, D. J. J. Am. Chem. Soc. 2016, 138, 5016–5019. doi:10.1021/jacs.6b01533 |
| 18. | Miyaura, N.; Yanagi, T.; Suzuki, A. Synth. Commun. 1981, 11, 513–519. doi:10.1080/00397918108063618 |
| 21. | Bloome, K. S.; McMahen, R. L.; Alexanian, E. J. J. Am. Chem. Soc. 2011, 133, 20146–20148. doi:10.1021/ja2091883 |
| 22. | Pearson, R. G.; Figdore, P. E. J. Am. Chem. Soc. 1980, 102, 1541–1547. doi:10.1021/ja00525a013 |
| 23. | Alisha, M.; Philip, R. M.; Anilkumar, G. J. Organomet. Chem. 2022, 959, 122207. doi:10.1016/j.jorganchem.2021.122207 |
| 14. | Frisch, A. C.; Beller, M. Angew. Chem., Int. Ed. 2005, 44, 674–688. doi:10.1002/anie.200461432 |
| 15. | Rudolph, A.; Lautens, M. Angew. Chem., Int. Ed. 2009, 48, 2656–2670. doi:10.1002/anie.200803611 |
| 16. | Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417–1492. doi:10.1021/cr100327p |
| 24. | Nishimura, T.; Uemura, S. J. Am. Chem. Soc. 2000, 122, 12049–12050. doi:10.1021/ja005558l |
| 25. | Nishimura, T.; Yoshinaka, T.; Nishiguchi, Y.; Maeda, Y.; Uemura, S. Org. Lett. 2005, 7, 2425–2427. doi:10.1021/ol0507120 |
| 26. | Yang, H.-B.; Selander, N. Chem. – Eur. J. 2017, 23, 1779–1783. doi:10.1002/chem.201605636 |
| 27. | Faulkner, A.; Race, N. J.; Scott, J. S.; Bower, J. F. Chem. Sci. 2014, 5, 2416–2421. doi:10.1039/c4sc00652f |
| 28. | Lin, X.; Stien, D.; Weinreb, S. M. Org. Lett. 1999, 1, 637–640. doi:10.1021/ol990720e |
| 29. | Krylov, I. B.; Segida, O. O.; Budnikov, A. S.; Terent'ev, A. O. Adv. Synth. Catal. 2021, 363, 2502–2528. doi:10.1002/adsc.202100058 |
| 11. | Jun, C.-H. Chem. Soc. Rev. 2004, 33, 610–618. doi:10.1039/b308864m |
| 12. | Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613–8661. doi:10.1021/cr400628s |
| 13. | Dermenci, A.; Coe, J. W.; Dong, G. Org. Chem. Front. 2014, 1, 567–581. doi:10.1039/c4qo00053f |
| 10. | Fleming, F. F.; Yao, L.; Ravikumar, P. C.; Funk, L.; Shook, B. C. J. Med. Chem. 2010, 53, 7902–7917. doi:10.1021/jm100762r |
| 14. | Frisch, A. C.; Beller, M. Angew. Chem., Int. Ed. 2005, 44, 674–688. doi:10.1002/anie.200461432 |
| 19. | Terao, J.; Kambe, N. Bull. Chem. Soc. Jpn. 2006, 79, 663–672. doi:10.1246/bcsj.79.663 |
| 20. | Sherry, B. D.; Fürstner, A. Acc. Chem. Res. 2008, 41, 1500–1511. doi:10.1021/ar800039x |
| 39. | Xiao, T.; Zhou, L.; Huang, H.; Anand, D. Synthesis 2020, 52, 1585–1601. doi:10.1055/s-0039-1690844 |
| 40. | Liu, L.; Duan, X.-H.; Guo, L.-N. Synthesis 2021, 53, 4375–4388. doi:10.1055/a-1545-6874 |
| 41. | Lu, X.-Y.; Xia, Z.-J.; Gao, A.; Liu, Q.-L.; Jiang, R.-C.; Liu, C.-C. J. Org. Chem. 2021, 86, 8829–8842. doi:10.1021/acs.joc.1c00726 |
| 36. | Gao, J.; Ye, Z.-P.; Liu, Y.-F.; He, X.-C.; Guan, J.-P.; Liu, F.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2022, 24, 4640–4644. doi:10.1021/acs.orglett.2c01750 |
| 37. | Zhao, J.-F.; Duan, X.-H.; Gu, Y.-R.; Gao, P.; Guo, L.-N. Org. Lett. 2018, 20, 4614–4617. doi:10.1021/acs.orglett.8b01901 |
| 38. | Gu, Y.-R.; Duan, X.-H.; Yang, L.; Guo, L.-N. Org. Lett. 2017, 19, 5908–5911. doi:10.1021/acs.orglett.7b02902 |
| 59. | Jiang, Y.; Sun, B.; Fang, W.-Y.; Qin, H.-L. Eur. J. Org. Chem. 2019, 3190–3194. doi:10.1002/ejoc.201900478 |
| 26. | Yang, H.-B.; Selander, N. Chem. – Eur. J. 2017, 23, 1779–1783. doi:10.1002/chem.201605636 |
| 30. | Zhao, B.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 12727–12731. doi:10.1002/anie.201707181 |
| 36. | Gao, J.; Ye, Z.-P.; Liu, Y.-F.; He, X.-C.; Guan, J.-P.; Liu, F.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2022, 24, 4640–4644. doi:10.1021/acs.orglett.2c01750 |
| 37. | Zhao, J.-F.; Duan, X.-H.; Gu, Y.-R.; Gao, P.; Guo, L.-N. Org. Lett. 2018, 20, 4614–4617. doi:10.1021/acs.orglett.8b01901 |
| 42. | Xia, P.-J.; Ye, Z.-P.; Hu, Y.-Z.; Song, D.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2019, 21, 2658–2662. doi:10.1021/acs.orglett.9b00651 |
| 60. | Li, L.; Chen, H.; Mei, M.; Zhou, L. Chem. Commun. 2017, 53, 11544–11547. doi:10.1039/c7cc07347j |
| 61. | Liu, X.-F.; Zhang, K.; Wang, L.-L.; Wang, H.; Huang, J.; Zhang, X.-T.; Lu, X.-B.; Zhang, W.-Z. J. Org. Chem. 2023, 88, 5212–5219. doi:10.1021/acs.joc.2c01816 |
| 44. | Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399 |
| 45. | Qin, H.-L.; Zheng, Q.; Bare, G. A. L.; Wu, P.; Sharpless, K. B. Angew. Chem., Int. Ed. 2016, 55, 14155–14158. doi:10.1002/anie.201608807 |
| 46. | Gao, B.; Zhang, L.; Zheng, Q.; Zhou, F.; Klivansky, L. M.; Lu, J.; Liu, Y.; Dong, J.; Wu, P.; Sharpless, K. B. Nat. Chem. 2017, 9, 1083–1088. doi:10.1038/nchem.2796 |
| 47. | Lekkala, R.; Lekkala, R.; Moku, B.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2019, 6, 3490–3516. doi:10.1039/c9qo00747d |
| 48. | Liu, J.; Wang, S.-M.; Qin, H.-L. Tetrahedron 2020, 76, 131724. doi:10.1016/j.tet.2020.131724 |
| 49. | Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a |
| 50. | Liu, J.; Wang, S.-M.; Alharbi, N. S.; Qin, H.-L. Beilstein J. Org. Chem. 2019, 15, 1907–1912. doi:10.3762/bjoc.15.186 |
| 51. | Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Alharbi, N. S.; Qin, H.-L. Tetrahedron 2019, 75, 4648–4656. doi:10.1016/j.tet.2019.07.007 |
| 52. | Fang, W.-Y.; Zha, G.-F.; Zhao, C.; Qin, H.-L. Chem. Commun. 2019, 55, 6273–6276. doi:10.1039/c9cc02659b |
| 53. | Wang, S.-M.; Alharbi, N. S.; Qin, H.-L. Synthesis 2019, 51, 3901–3907. doi:10.1055/s-0039-1690017 |
| 54. | Fang, W.-Y.; Qin, H.-L. J. Org. Chem. 2019, 84, 5803–5812. doi:10.1021/acs.joc.8b03164 |
| 55. | Epifanov, M.; Foth, P. J.; Gu, F.; Barrillon, C.; Kanani, S. S.; Higman, C. S.; Hein, J. E.; Sammis, G. M. J. Am. Chem. Soc. 2018, 140, 16464–16468. doi:10.1021/jacs.8b11309 |
| 56. | Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891 |
| 57. | Hanley, P. S.; Clark, T. P.; Krasovskiy, A. L.; Ober, M. S.; O’Brien, J. P.; Staton, T. S. ACS Catal. 2016, 6, 3515–3519. doi:10.1021/acscatal.6b00865 |
| 58. | Zhang, E.; Tang, J.; Li, S.; Wu, P.; Moses, J. E.; Sharpless, K. B. Chem. – Eur. J. 2016, 22, 5692–5697. doi:10.1002/chem.201600167 |
| 42. | Xia, P.-J.; Ye, Z.-P.; Hu, Y.-Z.; Song, D.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2019, 21, 2658–2662. doi:10.1021/acs.orglett.9b00651 |
| 43. | Sulbaek Andersen, M. P.; Blake, D. R.; Rowland, F. S.; Hurley, M. D.; Wallington, T. J. Environ. Sci. Technol. 2009, 43, 1067–1070. doi:10.1021/es802439f |
© 2023 Chen and Qin; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.