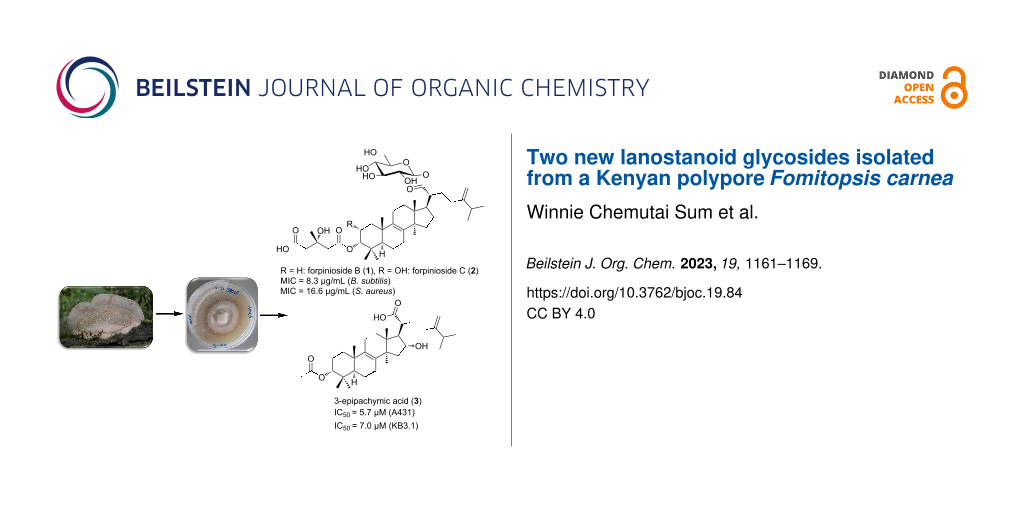
Abstract
Chemical exploration of solid-state cultures of the polypore Fomitopsis carnea afforded two new C31 lanostane-type triterpenoid glycosides, forpiniosides B (1) and C (2) together with two known derivatives, namely 3-epipachymic acid (3) and (3α,25S)-3-O-malonyl-23-oxolanost-8,24(31)-dien-26-oic acid (4). The structures of the isolated compounds were established based on HRESIMS and extensive 1D and 2D NMR experiments. All the isolated compounds were assessed for their antimicrobial and cytotoxic activities. Among the tested compounds, forpinioside B (1) exhibited significant antimicrobial activity against Staphylococcus aureus and Bacillus subtilis at MIC values comparable to gentamycin and oxytetracycline (positive controls), respectively.

Graphical Abstract
Introduction
Great success was realized on antibiotic discovery between 1930 and 1960 during the ‘golden era’ of antibiotics. Unfortunately, the pace of antibiotic research and development in the face of emerging resistant pathogens has not been kept up, thus raising a big concern for a return to the pre-antibiotic era [1,2]. Recently, the danger posed by previously treatable microbial diseases has been increased with the emergence of ‘superbugs’. If not properly addressed, these circumstances will inevitably lead to an increase in healthcare-associated costs due to longer and more frequent hospital stays as well as to the need of multi-drug therapy [3].
Due to their well-known profuse production of bioactive molecules, Basidiomycota have already been proven to be a valuable source for new anti-infectives [4]. These include a myriad of triterpenoids which have been resourceful in the discovery of potent antimicrobials [5]. Specifically, lanostane triterpenoids are typical bioactive chemical constituents of various Polyporales. For instance, these compounds play a major role in the pharmacological effects of Ganoderma such as Ganoderma lingzhi (often incorrectly referred by some scientists as "G. lucidum"), a medicinal fungus widely used in traditional Chinese medicine (TCM) [6]. Studies on other Ganoderma species have also reported antidiabetic [7] and cytotoxic [8] effects of these compounds. Another widely used oriental medicine polypore Wolfiporia (also referred to under the synonym ‘Poria’ in the literature) cocos, has proved to a beneficial source of bioactive lanostane triterpenoids [9,10]. In addition, the edible European fungus Macrolepiota procera also produced antiproliferative and anti-inflammatory lanostanoid derivatives [11]. Furthermore, triterpenes from Tricholoma pardinum and Fomitopsis betulina (previously known as Piptoporus betulinus) exhibited nitric oxide (NO) and/or cytotoxic effects, respectively [12,13].
The genus Fomitopsis was first coined by Karsten typified by F. pinicola [14]. Our current study fungus F. carnea was reported for the first time from Japan in 1943 by Blume and Nees [15]. Later, Ryvarden and Johansen in 1980 [16] and Carranza-Morse and Gilbertson in 1986 [17] confirmed the presence of the species in Tanzania apart from Japan. Ortiz-Santana et al. [18] indicated the close relationship of the genus Fomitopsis to Antrodia. This led to more recent revisions on the genera based on multiple genes (nLSU, ITS, nSSU, mtSSU, rbb2 and tef1), placing F. carnea in the Rhodofomes clade [15].
The genus Fomitopsis has proven to be an invaluable source of bioactive molecules [18-23]. Fruit body infusions of F. betulina have been widely used in folk medicine in combating various diseases and ailments [20]. Chemical investigations of F. pinicola or F. betulina revealed their cytotoxic [21,23] and antidiabetic propensities [19]. In addition, the recent review on F. officinalis, elaborated the potential use of its compounds as antibiotic leads [19].
In this study we report the isolation and structure elucidation of two new C31 lanostane-type triterpenoid glycosides (compounds 1 and 2 in Figure 1) together with two known derivatives, namely 3-epipachymic acid (3α-acetoxy-16α-hydroxy-5α-lanost-8,24(31)-dien-21-oic acid (3)) [24] and (3α,25S)-3-O-malonyl-23-oxolanost-8,24(31)-dien-26-oic acid (4) [25].
![[1860-5397-19-84-1]](/bjoc/content/figures/1860-5397-19-84-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of compounds 1–4.
Figure 1: Chemical structures of compounds 1–4.
Results
Structure elucidation
Compound 1 was isolated as an off-white solid powder. Its molecular formula was determined to be C43H68O12 based on HRESIMS results that revealed a sodium adduct ion peak at m/z 799.4604 ([M + Na]+ calcd for C43H68O12Na+, 799.4603) indicating the presence of ten degrees of unsaturation in its structure. The 1H, 13C NMR, and HSQC spectral data of compound 1 (Table 1) revealed the presence of forty-three carbon resonances sorted into eight methyl, fourteen methylenes (one olefinic), ten methine and eleven unprotonated carbon atoms. This includes three carbonyl carbons at δC 177.4 (C-21), 175.0 (C-5'), and 172.4 (C-1') as well as four olefinic carbon signals at δC 156.7 (C-24), 136.3 (C-9), 135.2 (C-8), and 107.4 (C-31). The 1H and 13C NMR spectral data of compound 1 (Table 1) also revealed the presence of a sugar moiety through the presence of the characteristic anomeric proton resonance at δH 5.49 (d, J = 8.1, H-1'') which was directly correlated to the carbon at δC 95.7 (C-1'') in the HSQC spectrum. The 1H,1H COSY spectrum of compound 1 (Figure 2) revealed an extended spin system over four aliphatic methines and ending with one aliphatic methylene. By comparing the 1H and 13C NMR data (Table 1) of the sugar moiety with that reported for a related fungal lanostanoside, ganosinoside A [26] and other glycosidic moieties [27], it was confirmed to be a β-ᴅ-glucopyranosyl residue. The HMBC spectrum of compound 1 (Figure 2) also revealed key correlations from two methylene groups at δH 2.65/2.69 (H2-2') and δH 2.69/2.72 (H2-4') to an ester carbonyl (δC 172.4, C-1') and a carboxyl (δC 175.0, C-5'), respectively. In addition, both methylene groups together with a methyl singlet at δH 1.38 (s, H3-6') disclosed key HMBC correlations to a quaternary oxygenated carbon at δC 70.7 (C-3') suggesting the presence of a 3-hydroxy-3-methylglutaroyl moiety in compound 1 [23].
Table 1: 1H and 13C NMR data of compounds 1 and 2 in methanol-d4.
| Pos. | 1 | 2 | ||
| δC,a,b type | δHc (multi, J (Hz)) | δC,a,b type | δHc (multi, J (Hz)) | |
| 1 | 32.1, CH2 | 1.51 (m, 2H) | 40.7, CH2 |
1.55 (t, 12.6, 1H)
1.73 (dd, 12.6, 4.0, 1H) |
| 2 | 24.2, CH2 |
1.65 (m, overlapped, 1H)
1.92 (m, overlapped, 1H) |
66.5, CH | 4.02 (ddd, 12.3, 4.3, 2.9, 1H) |
| 3 | 79.8, CH | 4.68 (t, 2.8, 1H) | 81.8, CH | 4.96 (dd, 2.9, 0.9, 1H) |
| 4 | 37.8, C | 39.0, C | ||
| 5 | 46.8, CH | 1.56 (br d, 2.0, 1H) | 46.2, CH | 1.46 (dd, 13.0, 2.2, 1H) |
| 6 | 19.1, CH2 |
1.56 (m, overlapped, 1H)
1.66 (m, overlapped, 1H) |
18.8, CH2 |
1.55 (m, overlapped, 1H)
1.66 (m, overlapped, 1H) |
| 7 | 27.1, CH2 | 1.41 (m, overlapped, 2H) | 29.8, CH2 |
1.54 (m, overlapped, 1H)
1.64 (m, overlapped, 1H) |
| 8 | 135.2, C | 135.3, C | ||
| 9 | 136.3, C | 136.0, C | ||
| 10 | 38.1, C | 39.5, C | ||
| 11 | 21.9, CH2 | 2.03 (m, overlapped, 2H) | 22.1, CH2 | 2.05 (m, overlapped, 2H) |
| 12 | 29.9, CH2 |
1.51 (m, overlapped, 1H)
1.61 (m, overlapped, 1H) |
29.8, CH2 |
1.53 (m, overlapped, 1H)
1.63 (m, overlapped, 1H) |
| 13 | 45.6, C | 45.6, C | ||
| 14 | 50.7, C | 50.6, C | ||
| 15 | 31.5, CH2 |
1.26 (m, overlapped, 1H)
1.67 (m, overlapped, 1H) |
31.5, CH2 |
1.27 (m, overlapped, 1H)
1.67 (m, overlapped, 1H) |
| 16 | 27.1, CH2 | 2.08 (m, overlapped, 2H) | 27.0, CH2 | 2.09 (m, overlapped, 2H) |
| 17 | 48.3, CH | 2.14 (m, 1H) | 48.3, CH | 2.14 (m, overlapped, 1H) |
| 18 | 16.7, CH3 | 0.81 (s, 3H) | 16.6, CH3 | 0.81 (s, 3H) |
| 19 | 19.5, CH3 | 1.03 (s, 3H) | 20.5, CH3 | 1.07 (s, 3H) |
| 20 | 48.8, CH | 2.40 (td, 11.0, 3.5, 1H) | 48.9, CH | 2.40 (td, 11.0, 3.4, 1H) |
| 21 | 177.4, C | 177.3, C | ||
| 22 | 32.6, CH2 |
1.68 (m, overlapped, 1H)
1.73 (m, overlapped, 1H) |
32.6, CH2 |
1.68 (m, overlapped, 1H)
1.73 (m, overlapped, 1H) |
| 23 | 32.7, CH2 |
1.98 (m, overlapped, 1H)
2.11 (m, overlapped, 1H) |
32.7, CH2 |
1.97 (m, overlapped, 1H)
2.12 (m, overlapped, 1H) |
| 24 | 156.7, C | 156.7, C | ||
| 25 | 35.0, CH | 2.23 (pd, 6.9, 1.1, 1H) | 35.0, CH | 2.24 (pd, 6.9, 1.0, 1H) |
| 26 | 22.3, CH3 | 1.03 (d, 6.8, 3H) | 22.3, CH3 | 1.03 (d, 6.9, 3H) |
| 27 | 22.4, CH3 | 1.01 (d, 6.8, 3H) | 22.4, CH3 | 1.01 (d, 6.9, 3H) |
| 28 | 28.4, CH3 | 0.90 (s, 3H) | 28.3, CH3 | 0.92 (s, 3H) |
| 29 | 22.3, CH3 | 0.95 (s, 3H) | 22.1, CH3 | 0.99 (s, 3H) |
| 30 | 24.7, CH3 | 0.94 (s, 3H) | 24.7, CH3 | 0.94 (s, 3H) |
| 31 | 107.4, CH2 |
4.70 (d, 1.4, 1H)
4.75 (d, 1.4, 1H) |
107.4, CH2 |
4.70 (d, 1.4, 1H)
4.75 (d, 1.4, 1H) |
| 1' | 172.4, C | 172.9, C | ||
| 2' | 46.5, CH2 |
2.65 (d, 15.1, 1H)
2.69 (d, 15.1, 1H) |
46.7, CH2 |
2.71 (d, 14.4, 1H)
2.77 (d, 14.4, 1H) |
| 3' | 70.7, C | 70.9, C | ||
| 4' | 45.9, CH2 |
2.69 (d, 14.9, 1H)
2.72 (d, 14.9, 1H) |
46.6, CH2 |
2.63 (d, 15.0, 1H)
2.66 (d, 15.0, 1H) |
| 5' | 175.0, C | 175.3, C | ||
| 6' | 27.9, CH3 | 1.38 (s, 3H) | 27.5, CH3 | 1.39 (s, 3H) |
| 1'' | 95.7, CH | 5.49 (d, 8.1, 1H) | 95.7, CH | 5.50 (d, 8.3, 1H) |
| 2'' | 73.9, CH | 3.35 (m, overlapped, 1H) | 73.9, CH | 3.36 (dd, 9.1, 8.2, 1H) |
| 3'' | 78.4, CH | 3.42 (m, overlapped, 1H) | 78.4, CH | 3.43 (d, 9.1, 1H) |
| 4'' | 71.2, CH | 3.39 (m, overlapped, 1H) | 71.2, CH | 3.39 (m, overlapped, 1H) |
| 5'' | 78.7, CH | 3.38 (m, overlapped, 1H) | 78.7, CH | 3.38 (m, overlapped, 1H) |
| 6'' | 62.6, CH2 |
3.71 (dd, 11.9, 1.8, 1H)
3.81 (dd, 11.8, 4.4, 1H) |
62.6, CH2 |
3.71 (dd, 11.8, 4.5, 1H)
3.81 (dd, 11.8, 1.8, 1H) |
aAt 500 MHz; bassigned based on HMBC and HSQC spectra; cat 125 MHz.
![[1860-5397-19-84-2]](/bjoc/content/figures/1860-5397-19-84-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Key 1H,1H COSY, HMBC, and ROESY correlations of compounds 1 and 2.
Figure 2: Key 1H,1H COSY, HMBC, and ROESY correlations of compounds 1 and 2.
These results illustrated that compound 1 is a C31 lanostane-type triterpenoid glucoside. According to the above results, the C31 lanostane-type triterpenoid moiety of compound 1 was identified to be a lanostan-8,24(31)-diene-21-oic acid skeleton supported by 2D NMR cross peaks in the 1H,1H COSY, HMBC, and HSQC spectra, suggesting a closely related structure to forpinioside A [23,28]. The C-5, C-10, C-13, and C-14 configurations were assigned not only from the biogenetic considerations, but also from careful comparison of the 1H and 13C NMR data with those of related compounds and ROESY correlations. The orientation of H-3 (δH 4.68, t, J = 2.8 Hz) was determined to be β based on comparing the measured and the reported coupling constants and chemical shifts for related lanostanoid derivatives [13,23,29,30].
Indeed, when the proton H-3 has a β-orientation as in compound 1, it resonates in the form of a broad singlet or else in the form of a triplet with a small coupling constant indicating its equatorial disposition [31,32]. When this proton has an α-orientation, it resonates as a doublet of doublet with coupling constants around 4.5 and 11 Hz [28,33]. The β-orientation of H-3 in compound 1 was further supported not only by the ROESY correlation observed between H-3 (δH 4.68, t, J = 2.8 Hz) and Me-29 (δH 0.95, s), but also by the fact that 3-epipachymic acid (3) and 3α,25S-3-O-malonyl-23-oxolanost-8,24(31)-dien-26-oic acid (4) were obtained during this investigation from the same extract. Some important ROESY correlations were depicted between Me-30 (δH 0.94, s) and H-17 (δH 2.14, m) as well as between H-20 (δH 2.40, td, J = 11.0, 3.5 Hz) and Me-18 (δH 0.81, s) indicating the α-orientation of H-17 and C-21, respectively. By comparing the HRESIMS, 1H and 13C NMR data of compound 1 and those reported for forpinioside A [23,28], it was obviously recognized that compound 1 lacks the ester group at δH 3.62 (OCH3-5'; δC 51.9) on the 3-hydroxy-3-methylglutaroyl substituent. Further confirmation of the positions of the 3-hydroxy-3-methylglutaroyl and β-ᴅ-glucopyranosyl moieties was provided by the HMBC spectrum which exhibited key correlations from H-3 to C-1' and from H-20 (δH 2.40, td, J = 11.0, 3.5 Hz)/H-1'' (δH 5.49, d, J = 8.1 Hz) to a carbonyl carbon at δC 177.4 (C-21). Hence, their positions were notably confirmed to be at C-3 and C-21, respectively. The ᴅ-configuration of the β-glucopyranosyl unit was assumed to be the one found in related fungal lanostanosides namely; ganosinoside A from Ganoderma sinense [26], fomitosides I and J from Fomitopsis pinicola [34]. Concerning the configuration at C-3', it was assigned as S by careful comparison of the 1H and 13C NMR data of compound 1 with those of related lanostane derivatives comprising the 3-hydroxy-3-methylglutaryl moiety produced by some fungi including Fomitopsis pinicola [23], Piptoporus betulinus [13], and Pholiota populnea [35]. Furthermore, it is well known that similar biosynthetic pathways might lead to the same product in most of the cases. Based on the aforementioned results, compound 1 was identified as 3α-[(3′S)-4′-carboxyl-3′-hydroxy-3′-methylbutanoyloxy]lanosta-8,24(31)-dien-21-oic acid 21-O-β-ᴅ-glucopyranoside that was trivially named as forpinioside B.
Compound 2 was obtained as a colourless oil and its molecular formula was determined to be C43H68O13 according to its HRESIMS spectrum that revealed a sodium adduct ion peak at m/z 815.4550 ([M + Na]+ calcd for C43H68O13Na+, 815.4552) indicating the presence of an additional oxygen atom compared to compound 1.
The 1H, 13C NMR, and HSQC spectral data of compound 2 (Table 1) revealed a close similarity to forpinioside B (1) apart from the presence of an additional aliphatic methine proton at δH 4.02 (ddd, J = 12.3, 4.3, 2.9 Hz; δC 66.5) which exhibited an obvious spin system in the 1H,1H COSY spectrum (Figure 2) with a methylene group at δH 1.51/δH 1.73 (H2-1) and a methine proton at δH 4.96 (H-3). These results suggested that compound 2 features a hydroxy group at C-2 rather than a methylene group as in forpinioside B (1). Further confirmation for the suggested position of the hydroxy group at C-2 was provided by the HMBC spectrum (Figure 2) which exhibited clear correlations from H-2 to four carbon resonances at δC 40.7 (C-1), 81.8 (C-3), 39.0 (C-4), and 39.5 (C-10).
Aside from this difference and by comparing the 1D and 2D NMR spectral data of compounds 1 and 2, they were closely related derivatives. The relative orientation of H-2 was determined to be β based on its ROESY spectrum (Figure 2) which revealed key NOE correlations to H-3 (δH 4.96) along with two singlet methyl groups namely, Me-19 (δH 1.07) and Me-29 (δH 0.99). Based on the above results, compound 2 was identified as 2α-hydroxy,3α-[(3′S)-4′-carboxyl-3′-hydroxy-3′-methylbutanoyloxy]lanosta-8,24(31)-dien-21-oic acid 21-O-β-ᴅ-glucopyranoside that was named as forpinioside C.
Biological activities
Compounds 1–4 were tested for their antimicrobial effects against fungi and bacteria; where compound 1 was moderately active against the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus at MIC values of 8.3 µg/mL and 16.6 µg/mL, respectively. The antagonism of 1 against B. subtilis and S. aureus was compared to the positive controls oxytetracycline and gentamycin, with MICs values recorded at 16.6 µg/mL and 0.21 µg/mL, respectively. Compounds 1, 2, and 4 exhibited no cytotoxic effects against highly sensitive mammalian cell lines namely; mouse fibroblasts (L929) and human endocervical adenocarcinoma cells (KB3.1), hence no further tests were made on the other cell lines (Table S1 in Supporting Information File 1). However, 3-epipachymic acid (3) demonstrated significant cytotoxicity against epidermoid carcinoma cells (A431) (IC50 = 5.7 µM) and HeLa cells (KB-3-1) (IC50 = 7.0 µM). Moderate cytotoxic effects for compound 3 were also recorded against mouse fibroblasts (L929) (IC50 = 15.2 µM), breast cancer cells (MCF-7) (17.6 µM), and prostate cancer cells (PC-3) (18.9 µM).
Discussion
The introduction of a hydroxy group at C-2 rendered forpinioside C (2) inactive in antimicrobial assays compared to forpinioside B (1), however; both compounds were not active in the cytotoxicity assay. Recent structure–activity relationship (SAR) studies have indicated a key role played by the hydroxy group at C-3 in cytotoxic effects of lanostane triterpenoids [23,36]. According to a study by Wang et al. (2023) [36], synthetic derivatives of pachymic acid demonstrated moderate to high potency against cancer cells in the presence of a 3-OH group. Notably, hydrolysis of the C3-acetoxy group in pachymic acid to tumulosic acid increased the activity of the compound compared to the positive control (cisplatin), in some instances [36]. Concomitantly, the oxidation of the hydroxy group at C-3 into a ketone moiety diminished the activity [31]. Similar findings on the related polyporenic acid B, with significant IC50 values between 8.4–12.2 µM [22], confirms the probable crucial role played by the 3-OH group in improving the potency. The presence of the 3-O-methylglutaroyl functionality in compound 1 seemed to be responsible for its antibiotic effects. The introduction of a 2-OH group in compound 2 could have interfered with its 3-O-methylglutaroyl group, rendering it inactive in the antimicrobial assay. However, as previous hinted by studies on similar derivatives [22], the cytotoxic effects of compound 1 and related analogues could be realized by C-3 hydrolysis.
In 2010, Liu et al. also demonstrated the major role played by changes at C-16 on antibacterial effects of F. pinicola steroids [33]. The acetylation at C-16 reduced the potency compared to the corresponding congener with a hydroxy group [33]. In our case, the C-16 hydroxy group seemed to play a major role in the cytotoxicity instead, as indicated by the active compound 3-epipachymic acid (3). Intriguingly, other compounds lacking the hydroxy group at C-16 were inactive, suggesting the probable role played by a 16-OH in inducing cytotoxicity. In addition, another study demonstrated that a lanostanoid glycoside derivative with a glucosyl ester at the C-21 carboxylic acid group was active in a cytotoxic activity assay, whereas the galactosyl ester counterpart was inactive [36]. Similarly, the carboxylic acid group at C-21 was shown to be a key player in lanostane triterpenoid cytotoxicity; increased activity was demonstrated either by its esterification with glucose or by its reduction into a hydroxymethylene group unlike its presence as a free carboxylic acid moiety [23]. Thus, our findings provide further insights into the SARs of lanostanoid triterpenoids and expands the database of their bioactive compounds for subsequent studies.
Conclusion
The genus Fomitopsis remains to be a prolific source of several metabolites with health-promoting effects. We provide a new evidence of lanostanoid glycosides 1 and 2 from solid-state cultivated cultures of F. carnea, with forpinioside B (1) proving to be a potential antimicrobial. In addition, our findings provide significant insights to decipher the SARs of the lanostanoid triterpenoids.
Experimental
General experimental methods
The samples were analyzed on an amaZon speed ETD ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) for HPLC-DAD/MS in positive and negative ionization modes. The HPLC (Dionex UltiMate 3000 UHPLC, Thermo Fisher Scientific Inc., Waltham, MA, USA) system’s stationary phase was composed of a C18 Acquity UPLC BEH column (Waters, Milford, MA, USA). The solvent system was as follows; deionized H2O + 0.1% formic acid (FA, v/v) (solvent A) and acetonitrile (ACN) + 0.1% FA (v/v) (solvent B). The separation gradient was operated as follows; 5% B for 0.5 min, 5% B to 100% B for 20 min, and 100% B for 10 min. The flow rate was maintained at 0.6 mL/min and the UV–vis detection made at 210 nm and 190–600 nm.
The isolates were analyzed on a MaXis ESI-TOF (time-of-flight) mass spectrometer (Bruker Daltonics) for the HRESIMS data, in the positive ionization mode. This was coupled to an Agilent 1260 series HPLC–UV system (Agilent Technologies, Santa Clara, CA, USA). The HPLC system’s stationary phase was composed of a C18 Acquity UPLC BEH column (Waters). The mobile phase employed was as follows; deionized H2O + 0.1% FA (v/v) (solvent A) and ACN + 0.1% FA (v/v) (solvent B). The separation gradient was operated as follows; 5% B for 0.5 min, 5% B to 100% B within 19.5 min and at 100% B for 5 min. The flow rate was maintained at 0.6 mL/min and UV–vis detection made at 200–600 nm. The calculation of the compounds’ molecular formulas was performed using the Smart Formula algorithm of the Compass DataAnalysis software (Bruker Daltonics, version 4.4 SR1).
The NMR spectra were recorded on an Avance III 700 (Bruker Biopsin, 1H: 700 MHz, 13C: 176 MHz) and/or an Avance III 500 (Bruker Biospin, Ettlingen, Germany, 1H: 500 MHz, 13C: 125 MHz) instruments. Deuterated methanol and dimethyl sulfoxide were used in the measurements. Coupling constants were reported in hertz (Hz) and chemical shifts in parts per million (ppm). The reference values 2.49 ppm and 3.31 ppm were used for the residual proton signals in the calibration of 1H NMR spectra for CD3SOCD3 and CD3OD, respectively. Likewise, the reference values of 39.5 and 49.1 for the deuterated solvents CD3SOCD3 and CD3OD, respectively, were used in 13C NMR spectra calibration. An Anton Paar MCP-150 Polarimeter (Graz, Austria), was used to measure the optical rotations on a 100 mm path length and sodium D line at 589 nm. The sample concentration was 1.0 mg/mL in MeOH. A Shimadzu UV–vis 2450 spectrophotometer (Kyoto, Japan) was used to measure the UV–vis spectra at a concentration of 0.02 mg/mL in MeOH.
The chemicals and solvents (analytical and HPLC grade) were purchased from Merck KGaA (Darmstadt, Germany), AppliChem GmbH (Darmstadt, Germany), Carl Roth GmbH & Co. KG (Karlsruhe, Germany), and Avantor Performance Materials (Deventer, Netherlands). An in-house Purelab® flex water purification system (Veolia Water Technologies, Celle, Germany), was used in the preparation of deionized water.
Fungal material examined
The current study specimen was collected by one of the authors C.D in Kakamega National Park, Kenya (0°17'3.19" N 34°45'8.24" E) from a dead tree trunk. A voucher specimen is deposited at the Mycothèque de la Université catholique de Louvain (MUCL), Belgium under designated number MUCL 56078. The fungal specimen was identified morphologically by author C.D by comparison of its morphological traits to close relatives.
Fermentation and metabolites extraction
Mycelial cultures of F. carnea were fermented on solid and liquid-state media based on previously established methodologies [37]. Essentially, the preparation of solid-state media was as follows: 90 mg of rice was weighed out into 10 × 500 mL Erlenmeyer flasks, each containing 90 mL of deionized H2O and autoclaved. In addition, 20 × 200 mL Erlenmeyer flasks containing cotton seed flour (Q6/2) liquid-state fermentation media were prepared as follows: media components (ᴅ-glucose 2.5 g/L, glycerol 10 g/L, cotton seed flour 5 g/L in distilled water), were mixed, pH adjusted to 7.2, and autoclaved. The media flasks were inoculated with 10 mycelial plugs (5 mm) each and incubated for 28 days, under shaking and static conditions for liquid and solid cultures, respectively. The cultures were extracted after incubation following the aforementioned established procedures to afford 0.5 g (mycelial) and 1.5 g (supernatant) ethyl acetate extracts for Q6/2 cultures and 3.44 g ethyl acetate extract for rice cultures.
Isolation of compounds 1–4
Purification of the rice and Q6/2 media cultures was achieved by using a reversed-phase HPLC system (PLC 2020; Gilson, Middleton, WI, USA), equipped with a VP Nucleodur 100-5 C-18 ec packed column (25 × 40 mm, 7 μm, Macherey-Nagel) as stationary phase. The liquid phase consisted of solvent A and solvent B. The elution gradients for the Q6/2 and rice extracts were identical and were performed as follows: isocratic conditions at 5% solvent B for 10 min, followed by an increase to 20% in 3 min, 20% to 90% within 50 min, 90% to 100% in 5 min and finally isocratic conditions of solvent B at 100% for 30 min. The flow rate in each case was 40 mL/min and UV detections were carried out at 210, 254, and 350 nm. The mycelial Q6/2 extract yielded compound 3 (6.4 mg, tR = 86–88 min) and the rice extract yielded compounds 4 (3.4 mg, tR = 53 min), 1 (13.3 mg, tR = 57–58 min), and 2 (4.4 mg, tR = 51 min).
Forpinioside B (1): off-white solid powder; ![[Graphic 1]](/bjoc/content/inline/1860-5397-19-84-i1.svg?max-width=637&scale=1.18182) +9 (c 0.1, MeOH); UV–vis (MeOH) λmax, nm (log ε): 196.5 (1.7); NMR data (1H NMR: 500 MHz, 13C NMR: 125 MHz in methanol-d4) see Table 1; HRMS–ESI (m/z): [M + Na]+ calcd for C43H68NaO12+, 799.4603; found, 799.4604; [2M + Na]+ calcd for C86H136NaO13+, 1575.9314; found, 1575.9321.
+9 (c 0.1, MeOH); UV–vis (MeOH) λmax, nm (log ε): 196.5 (1.7); NMR data (1H NMR: 500 MHz, 13C NMR: 125 MHz in methanol-d4) see Table 1; HRMS–ESI (m/z): [M + Na]+ calcd for C43H68NaO12+, 799.4603; found, 799.4604; [2M + Na]+ calcd for C86H136NaO13+, 1575.9314; found, 1575.9321.
Forpinioside C (2): pale yellow oil; ![[Graphic 2]](/bjoc/content/inline/1860-5397-19-84-i2.svg?max-width=637&scale=1.18182) +30 (c 0.1, MeOH); UV–vis (MeOH) λmax, nm (log ε): 196.5 (1.7); NMR data (1H NMR: 500 MHz, 13C NMR: 125 MHz in methanol-d4) see Table 1; HRMS–ESI (m/z): [M – H2O + H]+ calcd for C43H67O12+, 775.4627; found, 775.4625; [M + Na]+ calcd for C43H68NaO13+, 815.4552; found, 815.4550; [2M + H]+ calcd for C86H137O26+, 1607.9212; found, 1607.9210.
+30 (c 0.1, MeOH); UV–vis (MeOH) λmax, nm (log ε): 196.5 (1.7); NMR data (1H NMR: 500 MHz, 13C NMR: 125 MHz in methanol-d4) see Table 1; HRMS–ESI (m/z): [M – H2O + H]+ calcd for C43H67O12+, 775.4627; found, 775.4625; [M + Na]+ calcd for C43H68NaO13+, 815.4552; found, 815.4550; [2M + H]+ calcd for C86H137O26+, 1607.9212; found, 1607.9210.
Antimicrobial assays
A wide array of microbial test pathogens were used for the antimicrobial assays according to our previously laid-out protocol [38]. The minimum inhibitory concentrations (MICs) of the compounds were determined via serial dilutions on 96-well microtiter plates. In brief, 20 µL aliquots of the compounds (1 mg/mL), in methanol were pipetted into the 96-well plates consisting of the respective test microorganisms, with a concentration range between 67 µg/mL and 0.5 µg/mL.
Cytotoxicity assays
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) test was used in determining the cytotoxicity (IC50) of the isolated compounds as previously established [38,39]. The mammalian cell lines (mouse fibroblasts L929, adenocarcinomic human alveolar basal epithelial cells A549, HeLa cells KB-3-1, breast cancer cells MCF-7, epidermoid carcinoma cells (A431), and prostate cancer cells PC-3) were obtained from the DSMZ collection (Braunschweig, Germany).
Supporting Information
| Supporting Information File 1: HRESIMS profiles and NMR spectroscopic data of 1, 2 and 4 in CD3OD, and of 3 in (CD3)2S=O; half inhibitory concentrations (IC50) for various mammalian cell lines as well as minimum inhibitory concentrations (MIC) of 1–4 for bacteria, yeasts and filamentous fungi. | ||
| Format: PDF | Size: 5.8 MB | Download |
Acknowledgements
The technical support accorded to this work by W. Collisi, A. Gollasch, E. Surges, C. Kakoschke, and K. Harmrolfs is greatly appreciated.
Funding
Our research benefitted from funding by the European Union’s Horizon 2020 research and innovation program (RISE) under the Marie Skłodowska‐Curie grant agreement No. 101008129, project acronym “Mycobiomics” (lead beneficiaries J.C.M. and M.S.). W.C.S. was supported by a doctoral scholarship funding from the German Academic Exchange Service (DAAD) program number 57507871. In addition, the financial grant of the Alexander von Humboldt Foundation (AvH) (Bonn, 3.4-CMR-Hub) and AvH Georg-Forster Fellowship for Experienced Researchers stipend (Ref 3.4-1222288-EGY-GF-E) to S.S.E. is acknowledged.
References
-
Aslam, B.; Wang, W.; Arshad, M. I.; Khurshid, M.; Muzammil, S.; Rasool, M. H.; Nisar, M. A.; Alvi, R. F.; Aslam, M. A.; Qamar, M. U.; Salamat, M. K. F.; Baloch, Z. Infect. Drug Resist. 2018, 11, 1645–1658. doi:10.2147/idr.s173867
Return to citation in text: [1] -
Brown, E. D.; Wright, G. D. Nature 2016, 529, 336–343. doi:10.1038/nature17042
Return to citation in text: [1] -
Ventola, C. L. Pharm. Ther. 2015, 40, 277.
Return to citation in text: [1] -
Sandargo, B.; Chepkirui, C.; Cheng, T.; Chaverra-Muñoz, L.; Thongbai, B.; Stadler, M.; Hüttel, S. Biotechnol. Adv. 2019, 37, 107344. doi:10.1016/j.biotechadv.2019.01.011
Return to citation in text: [1] -
Zhou, M.; Zhang, R.-H.; Wang, M.; Xu, G.-B.; Liao, S.-G. Eur. J. Med. Chem. 2017, 131, 222–236. doi:10.1016/j.ejmech.2017.03.005
Return to citation in text: [1] -
Su, H.-G.; Peng, X.-R.; Shi, Q.-Q.; Huang, Y.-J.; Zhou, L.; Qiu, M.-H. Phytochemistry 2020, 173, 112256. doi:10.1016/j.phytochem.2019.112256
Return to citation in text: [1] -
Yang, L.; Kong, D.-X.; Xiao, N.; Ma, Q.-Y.; Xie, Q.-Y.; Guo, J.-C.; Ying Deng, C.; Ma, H.-X.; Hua, Y.; Dai, H.-F.; Zhao, Y.-X. Bioorg. Chem. 2022, 127, 106025. doi:10.1016/j.bioorg.2022.106025
Return to citation in text: [1] -
Su, H.-G.; Zhou, Q.-M.; Guo, L.; Huang, Y.-J.; Peng, C.; Xiong, L. Phytochemistry 2018, 156, 89–95. doi:10.1016/j.phytochem.2018.09.003
Return to citation in text: [1] -
Chao, C.-L.; Huang, H.-W.; Su, M.-H.; Lin, H.-C.; Wu, W.-M. Life 2021, 11, 111. doi:10.3390/life11020111
Return to citation in text: [1] -
Lee, S.; Lee, D.; Lee, S. O.; Ryu, J.-Y.; Choi, S.-Z.; Kang, K. S.; Kim, K. H. J. Funct. Foods 2017, 32, 27–36. doi:10.1016/j.jff.2017.02.012
Return to citation in text: [1] -
Chen, H.-P.; Zhao, Z.-Z.; Li, Z.-H.; Huang, Y.; Zhang, S.-B.; Tang, Y.; Yao, J.-N.; Chen, L.; Isaka, M.; Feng, T.; Liu, J.-K. J. Agric. Food Chem. 2018, 66, 3146–3154. doi:10.1021/acs.jafc.8b00287
Return to citation in text: [1] -
Zhang, S.-B.; Li, Z.-H.; Stadler, M.; Chen, H.-P.; Huang, Y.; Gan, X.-Q.; Feng, T.; Liu, J.-K. Phytochemistry 2018, 152, 105–112. doi:10.1016/j.phytochem.2018.05.002
Return to citation in text: [1] -
Tohtahon, Z.; Xue, J.; Han, J.; Liu, Y.; Hua, H.; Yuan, T. Phytochemistry 2017, 143, 98–103. doi:10.1016/j.phytochem.2017.07.013
Return to citation in text: [1] [2] [3] -
Karsten, P. A. Meddelanden Soc. Fauna Flora Fenn. 1879, 5, 15–46.
Return to citation in text: [1] -
Han, M.-L.; Chen, Y.-Y.; Shen, L.-L.; Song, J.; Vlasák, J.; Dai, Y.-C.; Cui, B.-K. Fungal Diversity 2016, 80, 343–373. doi:10.1007/s13225-016-0364-y
Return to citation in text: [1] [2] -
Ryvarden, L.; Johansen, I. A Preliminary Polypore Flora of East Africa; Cornell University: New York, NY, USA, 1980.
Return to citation in text: [1] -
Carranza-Morse, L.; Gilbertson, R. Mycotaxon 1986, 25, 469–486.
Return to citation in text: [1] -
Ortiz-Santana, B.; Lindner, D. L.; Miettinen, O.; Justo, A.; Hibbett, D. S. Mycologia 2013, 105, 1391–1411. doi:10.3852/13-051
Return to citation in text: [1] [2] -
Zhang, J.; Chen, B.; Liang, J.; Han, J.; Zhou, L.; Zhao, R.; Liu, H.; Dai, H. J. Agric. Food Chem. 2020, 68, 10036–10049. doi:10.1021/acs.jafc.0c04460
Return to citation in text: [1] [2] [3] -
Verekar, S. A.; Gupta, M. K.; Deshmukh, S. K. Fomitopsis betulina A Rich Source of Diverse Bioactive Metabolites. Advances in Macrofungi, 1st ed.; CRC Press: Boca Raton, FL, USA, 2021; pp 52–66. doi:10.1201/9781003191278-5
Return to citation in text: [1] [2] -
Sofrenić, I.; Anđelković, B.; Todorović, N.; Stanojković, T.; Vujisić, L.; Novaković, M.; Milosavljević, S.; Tešević, V. Phytochemistry 2021, 181, 112580. doi:10.1016/j.phytochem.2020.112580
Return to citation in text: [1] [2] -
Zhao, J.; Yang, Y.; Yu, M.; Yao, K.; Luo, X.; Qi, H.; Zhang, G.; Luo, Y. Phytochemistry 2018, 152, 10–21. doi:10.1016/j.phytochem.2018.04.012
Return to citation in text: [1] [2] [3] -
Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Niu, X.-M.; Li, S.-H.; Xiao, W.-L.; Sun, H.-D.; Che, C.-T. J. Asian Nat. Prod. Res. 2007, 9, 659–664. doi:10.1080/10286020600979910
Return to citation in text: [1] -
Chairul; Tokuyama, T.; Nishizawa, M.; Shiro, M.; Tokuda, H.; Hayashiji, Y. Phytochemistry 1990, 29, 923–928. doi:10.1016/0031-9422(90)80047-k
Return to citation in text: [1] -
Liu, J.-Q.; Wang, C.-F.; Li, Y.; Luo, H.-R.; Qiu, M.-H. Planta Med. 2012, 78, 368–376. doi:10.1055/s-0031-1280441
Return to citation in text: [1] [2] -
Agrawal, P. K. Phytochemistry 1992, 31, 3307–3330. doi:10.1016/0031-9422(92)83678-r
Return to citation in text: [1] -
Keller, A. C.; Maillard, M. P.; Hostettmann, K. Phytochemistry 1996, 41, 1041–1046. doi:10.1016/0031-9422(95)00762-8
Return to citation in text: [1] [2] [3] -
Zhou, L.; Zhang, Y.; Gapter, L. A.; Ling, H.; Agarwal, R.; Ng, K.-y. Chem. Pharm. Bull. 2008, 56, 1459–1462. doi:10.1248/cpb.56.1459
Return to citation in text: [1] -
Kamo, T.; Asanoma, M.; Shibata, H.; Hirota, M. J. Nat. Prod. 2003, 66, 1104–1106. doi:10.1021/np0300479
Return to citation in text: [1] -
Lin, L.-J.; Shiao, M.-S.; Yeh, S.-F. J. Nat. Prod. 1988, 51, 918–924. doi:10.1021/np50059a017
Return to citation in text: [1] [2] -
de Silva, E. D.; van der Sar, S. A.; Santha, R. G. L.; Wijesundera, R. L. C.; Cole, A. L. J.; Blunt, J. W.; Munro, M. H. G. J. Nat. Prod. 2006, 69, 1245–1248. doi:10.1021/np0602214
Return to citation in text: [1] -
Liu, X.-T.; Winkler, A. L.; Schwan, W. R.; Volk, T. J.; Rott, M.; Monte, A. Planta Med. 2010, 76, 464–466. doi:10.1055/s-0029-1186227
Return to citation in text: [1] [2] [3] -
Yoshikawa, K.; Inoue, M.; Matsumoto, Y.; Sakakibara, C.; Miyataka, H.; Matsumoto, H.; Arihara, S. J. Nat. Prod. 2005, 68, 69–73. doi:10.1021/np040130b
Return to citation in text: [1] -
Yazdani, M.; Béni, Z.; Dékány, M.; Szemerédi, N.; Spengler, G.; Hohmann, J.; Ványolós, A. J. Nat. Prod. 2022, 85, 910–916. doi:10.1021/acs.jnatprod.1c01024
Return to citation in text: [1] -
Wang, H.; Sun, X.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L. Med. Chem. Res. 2023, 32, 342–354. doi:10.1007/s00044-022-03009-3
Return to citation in text: [1] [2] [3] [4] -
Chepkirui, C.; Cheng, T.; Sum, W. C.; Matasyoh, J. C.; Decock, C.; Praditya, D. F.; Wittstein, K.; Steinmann, E.; Stadler, M. J. Agric. Food Chem. 2019, 67, 8468–8475. doi:10.1021/acs.jafc.9b02598
Return to citation in text: [1] -
Sum, W. C.; Mitschke, N.; Schrey, H.; Wittstein, K.; Kellner, H.; Stadler, M.; Matasyoh, J. C. Antibiotics (Basel, Switz.) 2022, 11, 1072. doi:10.3390/antibiotics11081072
Return to citation in text: [1] [2] -
Becker, K.; Wessel, A.-C.; Luangsa-ard, J. J.; Stadler, M. Biomolecules 2020, 10, 805. doi:10.3390/biom10050805
Return to citation in text: [1]
| 13. | Tohtahon, Z.; Xue, J.; Han, J.; Liu, Y.; Hua, H.; Yuan, T. Phytochemistry 2017, 143, 98–103. doi:10.1016/j.phytochem.2017.07.013 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 29. | Zhou, L.; Zhang, Y.; Gapter, L. A.; Ling, H.; Agarwal, R.; Ng, K.-y. Chem. Pharm. Bull. 2008, 56, 1459–1462. doi:10.1248/cpb.56.1459 |
| 30. | Kamo, T.; Asanoma, M.; Shibata, H.; Hirota, M. J. Nat. Prod. 2003, 66, 1104–1106. doi:10.1021/np0300479 |
| 31. | Lin, L.-J.; Shiao, M.-S.; Yeh, S.-F. J. Nat. Prod. 1988, 51, 918–924. doi:10.1021/np50059a017 |
| 32. | de Silva, E. D.; van der Sar, S. A.; Santha, R. G. L.; Wijesundera, R. L. C.; Cole, A. L. J.; Blunt, J. W.; Munro, M. H. G. J. Nat. Prod. 2006, 69, 1245–1248. doi:10.1021/np0602214 |
| 28. | Keller, A. C.; Maillard, M. P.; Hostettmann, K. Phytochemistry 1996, 41, 1041–1046. doi:10.1016/0031-9422(95)00762-8 |
| 33. | Liu, X.-T.; Winkler, A. L.; Schwan, W. R.; Volk, T. J.; Rott, M.; Monte, A. Planta Med. 2010, 76, 464–466. doi:10.1055/s-0029-1186227 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 36. | Wang, H.; Sun, X.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L. Med. Chem. Res. 2023, 32, 342–354. doi:10.1007/s00044-022-03009-3 |
| 36. | Wang, H.; Sun, X.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L. Med. Chem. Res. 2023, 32, 342–354. doi:10.1007/s00044-022-03009-3 |
| 13. | Tohtahon, Z.; Xue, J.; Han, J.; Liu, Y.; Hua, H.; Yuan, T. Phytochemistry 2017, 143, 98–103. doi:10.1016/j.phytochem.2017.07.013 |
| 35. | Yazdani, M.; Béni, Z.; Dékány, M.; Szemerédi, N.; Spengler, G.; Hohmann, J.; Ványolós, A. J. Nat. Prod. 2022, 85, 910–916. doi:10.1021/acs.jnatprod.1c01024 |
| 34. | Yoshikawa, K.; Inoue, M.; Matsumoto, Y.; Sakakibara, C.; Miyataka, H.; Matsumoto, H.; Arihara, S. J. Nat. Prod. 2005, 68, 69–73. doi:10.1021/np040130b |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 28. | Keller, A. C.; Maillard, M. P.; Hostettmann, K. Phytochemistry 1996, 41, 1041–1046. doi:10.1016/0031-9422(95)00762-8 |
| 26. | Liu, J.-Q.; Wang, C.-F.; Li, Y.; Luo, H.-R.; Qiu, M.-H. Planta Med. 2012, 78, 368–376. doi:10.1055/s-0031-1280441 |
| 36. | Wang, H.; Sun, X.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L. Med. Chem. Res. 2023, 32, 342–354. doi:10.1007/s00044-022-03009-3 |
| 31. | Lin, L.-J.; Shiao, M.-S.; Yeh, S.-F. J. Nat. Prod. 1988, 51, 918–924. doi:10.1021/np50059a017 |
| 22. | Zhao, J.; Yang, Y.; Yu, M.; Yao, K.; Luo, X.; Qi, H.; Zhang, G.; Luo, Y. Phytochemistry 2018, 152, 10–21. doi:10.1016/j.phytochem.2018.04.012 |
| 38. | Sum, W. C.; Mitschke, N.; Schrey, H.; Wittstein, K.; Kellner, H.; Stadler, M.; Matasyoh, J. C. Antibiotics (Basel, Switz.) 2022, 11, 1072. doi:10.3390/antibiotics11081072 |
| 38. | Sum, W. C.; Mitschke, N.; Schrey, H.; Wittstein, K.; Kellner, H.; Stadler, M.; Matasyoh, J. C. Antibiotics (Basel, Switz.) 2022, 11, 1072. doi:10.3390/antibiotics11081072 |
| 39. | Becker, K.; Wessel, A.-C.; Luangsa-ard, J. J.; Stadler, M. Biomolecules 2020, 10, 805. doi:10.3390/biom10050805 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 37. | Chepkirui, C.; Cheng, T.; Sum, W. C.; Matasyoh, J. C.; Decock, C.; Praditya, D. F.; Wittstein, K.; Steinmann, E.; Stadler, M. J. Agric. Food Chem. 2019, 67, 8468–8475. doi:10.1021/acs.jafc.9b02598 |
| 33. | Liu, X.-T.; Winkler, A. L.; Schwan, W. R.; Volk, T. J.; Rott, M.; Monte, A. Planta Med. 2010, 76, 464–466. doi:10.1055/s-0029-1186227 |
| 36. | Wang, H.; Sun, X.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L. Med. Chem. Res. 2023, 32, 342–354. doi:10.1007/s00044-022-03009-3 |
| 22. | Zhao, J.; Yang, Y.; Yu, M.; Yao, K.; Luo, X.; Qi, H.; Zhang, G.; Luo, Y. Phytochemistry 2018, 152, 10–21. doi:10.1016/j.phytochem.2018.04.012 |
| 33. | Liu, X.-T.; Winkler, A. L.; Schwan, W. R.; Volk, T. J.; Rott, M.; Monte, A. Planta Med. 2010, 76, 464–466. doi:10.1055/s-0029-1186227 |
| 1. | Aslam, B.; Wang, W.; Arshad, M. I.; Khurshid, M.; Muzammil, S.; Rasool, M. H.; Nisar, M. A.; Alvi, R. F.; Aslam, M. A.; Qamar, M. U.; Salamat, M. K. F.; Baloch, Z. Infect. Drug Resist. 2018, 11, 1645–1658. doi:10.2147/idr.s173867 |
| 2. | Brown, E. D.; Wright, G. D. Nature 2016, 529, 336–343. doi:10.1038/nature17042 |
| 6. | Su, H.-G.; Peng, X.-R.; Shi, Q.-Q.; Huang, Y.-J.; Zhou, L.; Qiu, M.-H. Phytochemistry 2020, 173, 112256. doi:10.1016/j.phytochem.2019.112256 |
| 18. | Ortiz-Santana, B.; Lindner, D. L.; Miettinen, O.; Justo, A.; Hibbett, D. S. Mycologia 2013, 105, 1391–1411. doi:10.3852/13-051 |
| 5. | Zhou, M.; Zhang, R.-H.; Wang, M.; Xu, G.-B.; Liao, S.-G. Eur. J. Med. Chem. 2017, 131, 222–236. doi:10.1016/j.ejmech.2017.03.005 |
| 15. | Han, M.-L.; Chen, Y.-Y.; Shen, L.-L.; Song, J.; Vlasák, J.; Dai, Y.-C.; Cui, B.-K. Fungal Diversity 2016, 80, 343–373. doi:10.1007/s13225-016-0364-y |
| 4. | Sandargo, B.; Chepkirui, C.; Cheng, T.; Chaverra-Muñoz, L.; Thongbai, B.; Stadler, M.; Hüttel, S. Biotechnol. Adv. 2019, 37, 107344. doi:10.1016/j.biotechadv.2019.01.011 |
| 16. | Ryvarden, L.; Johansen, I. A Preliminary Polypore Flora of East Africa; Cornell University: New York, NY, USA, 1980. |
| 11. | Chen, H.-P.; Zhao, Z.-Z.; Li, Z.-H.; Huang, Y.; Zhang, S.-B.; Tang, Y.; Yao, J.-N.; Chen, L.; Isaka, M.; Feng, T.; Liu, J.-K. J. Agric. Food Chem. 2018, 66, 3146–3154. doi:10.1021/acs.jafc.8b00287 |
| 9. | Chao, C.-L.; Huang, H.-W.; Su, M.-H.; Lin, H.-C.; Wu, W.-M. Life 2021, 11, 111. doi:10.3390/life11020111 |
| 10. | Lee, S.; Lee, D.; Lee, S. O.; Ryu, J.-Y.; Choi, S.-Z.; Kang, K. S.; Kim, K. H. J. Funct. Foods 2017, 32, 27–36. doi:10.1016/j.jff.2017.02.012 |
| 15. | Han, M.-L.; Chen, Y.-Y.; Shen, L.-L.; Song, J.; Vlasák, J.; Dai, Y.-C.; Cui, B.-K. Fungal Diversity 2016, 80, 343–373. doi:10.1007/s13225-016-0364-y |
| 8. | Su, H.-G.; Zhou, Q.-M.; Guo, L.; Huang, Y.-J.; Peng, C.; Xiong, L. Phytochemistry 2018, 156, 89–95. doi:10.1016/j.phytochem.2018.09.003 |
| 7. | Yang, L.; Kong, D.-X.; Xiao, N.; Ma, Q.-Y.; Xie, Q.-Y.; Guo, J.-C.; Ying Deng, C.; Ma, H.-X.; Hua, Y.; Dai, H.-F.; Zhao, Y.-X. Bioorg. Chem. 2022, 127, 106025. doi:10.1016/j.bioorg.2022.106025 |
| 12. | Zhang, S.-B.; Li, Z.-H.; Stadler, M.; Chen, H.-P.; Huang, Y.; Gan, X.-Q.; Feng, T.; Liu, J.-K. Phytochemistry 2018, 152, 105–112. doi:10.1016/j.phytochem.2018.05.002 |
| 13. | Tohtahon, Z.; Xue, J.; Han, J.; Liu, Y.; Hua, H.; Yuan, T. Phytochemistry 2017, 143, 98–103. doi:10.1016/j.phytochem.2017.07.013 |
| 21. | Sofrenić, I.; Anđelković, B.; Todorović, N.; Stanojković, T.; Vujisić, L.; Novaković, M.; Milosavljević, S.; Tešević, V. Phytochemistry 2021, 181, 112580. doi:10.1016/j.phytochem.2020.112580 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 18. | Ortiz-Santana, B.; Lindner, D. L.; Miettinen, O.; Justo, A.; Hibbett, D. S. Mycologia 2013, 105, 1391–1411. doi:10.3852/13-051 |
| 19. | Zhang, J.; Chen, B.; Liang, J.; Han, J.; Zhou, L.; Zhao, R.; Liu, H.; Dai, H. J. Agric. Food Chem. 2020, 68, 10036–10049. doi:10.1021/acs.jafc.0c04460 |
| 20. | Verekar, S. A.; Gupta, M. K.; Deshmukh, S. K. Fomitopsis betulina A Rich Source of Diverse Bioactive Metabolites. Advances in Macrofungi, 1st ed.; CRC Press: Boca Raton, FL, USA, 2021; pp 52–66. doi:10.1201/9781003191278-5 |
| 21. | Sofrenić, I.; Anđelković, B.; Todorović, N.; Stanojković, T.; Vujisić, L.; Novaković, M.; Milosavljević, S.; Tešević, V. Phytochemistry 2021, 181, 112580. doi:10.1016/j.phytochem.2020.112580 |
| 22. | Zhao, J.; Yang, Y.; Yu, M.; Yao, K.; Luo, X.; Qi, H.; Zhang, G.; Luo, Y. Phytochemistry 2018, 152, 10–21. doi:10.1016/j.phytochem.2018.04.012 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 20. | Verekar, S. A.; Gupta, M. K.; Deshmukh, S. K. Fomitopsis betulina A Rich Source of Diverse Bioactive Metabolites. Advances in Macrofungi, 1st ed.; CRC Press: Boca Raton, FL, USA, 2021; pp 52–66. doi:10.1201/9781003191278-5 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 23. | Peng, X.-R.; Su, H.-G.; Liu, J.-H.; Huang, Y.-J.; Yang, X.-Z.; Li, Z.-R.; Zhou, L.; Qiu, M.-H. J. Agric. Food Chem. 2019, 67, 10330–10341. doi:10.1021/acs.jafc.9b04530 |
| 28. | Keller, A. C.; Maillard, M. P.; Hostettmann, K. Phytochemistry 1996, 41, 1041–1046. doi:10.1016/0031-9422(95)00762-8 |
| 26. | Liu, J.-Q.; Wang, C.-F.; Li, Y.; Luo, H.-R.; Qiu, M.-H. Planta Med. 2012, 78, 368–376. doi:10.1055/s-0031-1280441 |
| 27. | Agrawal, P. K. Phytochemistry 1992, 31, 3307–3330. doi:10.1016/0031-9422(92)83678-r |
| 24. | Niu, X.-M.; Li, S.-H.; Xiao, W.-L.; Sun, H.-D.; Che, C.-T. J. Asian Nat. Prod. Res. 2007, 9, 659–664. doi:10.1080/10286020600979910 |
| 25. | Chairul; Tokuyama, T.; Nishizawa, M.; Shiro, M.; Tokuda, H.; Hayashiji, Y. Phytochemistry 1990, 29, 923–928. doi:10.1016/0031-9422(90)80047-k |
| 19. | Zhang, J.; Chen, B.; Liang, J.; Han, J.; Zhou, L.; Zhao, R.; Liu, H.; Dai, H. J. Agric. Food Chem. 2020, 68, 10036–10049. doi:10.1021/acs.jafc.0c04460 |
| 19. | Zhang, J.; Chen, B.; Liang, J.; Han, J.; Zhou, L.; Zhao, R.; Liu, H.; Dai, H. J. Agric. Food Chem. 2020, 68, 10036–10049. doi:10.1021/acs.jafc.0c04460 |
© 2023 Sum et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.