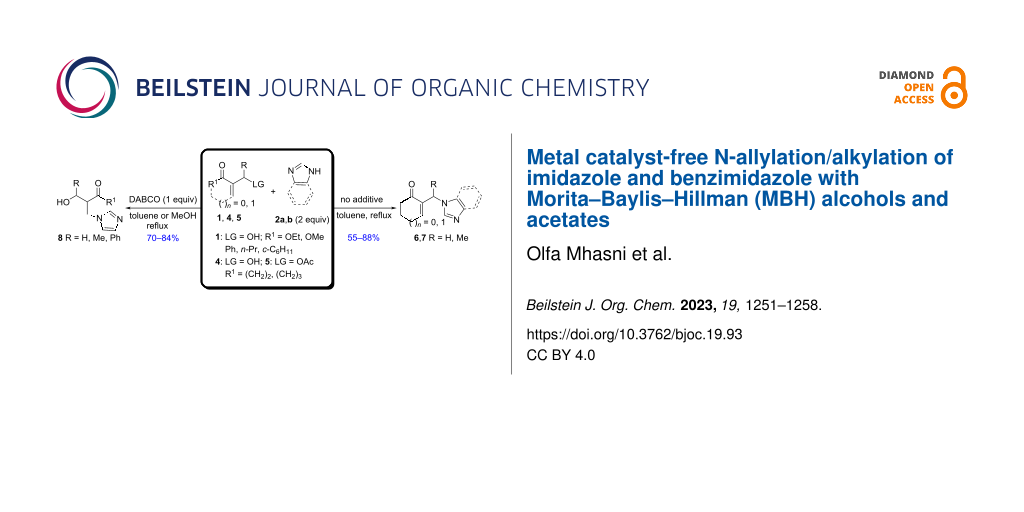
Abstract
A highly α-regioselective N-nucleophilic allylic substitution of cyclic MBH alcohols and acetates with imidazole or benzimidazole, in toluene at reflux with an azeotropic distillation, was successfully carried out with no catalysts or additives, affording the corresponding N-substituted imidazole derivatives in good yields. On the other hand, in refluxing toluene or methanol, the aza-Michael addition of imidazole onto acyclic MBH alcohols was performed using DABCO as an additive, leading to the corresponding 1,4-adducts in 70–84% yields.

Graphical Abstract
Introduction
Morita–Baylis–Hillman (MBH) adducts are multifunctionalized compounds having both a hydroxy moiety and a Michael acceptor unit. They have found application as valuable synthons and useful precursors for the synthesis of various biologically active molecules [1-3]. Recently, MBH adducts, as electrophilic substrates, have been employed to achieve fruitful results in allylic substitution reactions with various nucleophiles, including C- and heteronucleophiles, such as compounds bearing –OH, –SH, and –NH groups [4-7]. Among them, the carbon–nitrogen bond formation through N-nucleophilic substitution reactions plays a central role for the synthesis of numerous compounds exhibiting various biological activities [1-7].
In this context, the imidazole moiety is widely known as one of the most important groups which plays efficient roles in bioactive compounds [8]. For instance, a number of N-substituted imidazole derivatives, such as miconazole, ketoconazole, genaconazole, and bifonazole have become well-established drugs for the treatment of numerous mycotic infections (Figure 1) [9,10]. Therefore, the development of new methods for the preparation of such compounds is highly required.
![[1860-5397-19-93-1]](/bjoc/content/figures/1860-5397-19-93-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Medicines containing an imidazole nucleus.
Figure 1: Medicines containing an imidazole nucleus.
The MBH acetates, instead of the corresponding alcohols, have been extensively used as precursors in nucleophilic allylic substitution reactions with amines, presumably due to the perceived poor leaving group ability and low reactivity of the hydroxy group. Interestingly, the direct nucleophilic substitution of the corresponding alcohols has drawn much attention because of the availability of these substrates and the formation of water as the sole non-toxic byproduct in the reaction [11]. In general, the previous methods for the amination of MBH alcohols needed catalysts or additives such as FeCl3 [12,13], In(OTf)3 [14], MoCl5 [15], AuCl3 [16], and I2 [17] as Lewis acids. Alternatively, Yang et al. [18,19] have developed a catalytic system involving Pd/Ti(OiPr)4 or Pd/carboxylic acid for the direct allylation of anilines with alcohols.
The synthesis of N-allylimidazole derivatives 3 has been previously carried out using acyclic MBH adducts bearing good leaving groups, such as bromide derivatives in Et3N/THF mixtures [20] (Scheme 1, reaction 1, i) and acetates in THF/water [21] (Scheme 1, reaction 1, ii), or MBH alcohols in the presence of CDI (1,1’-carbonyldiimidazole) in acetonitrile (Scheme 1, reaction 1, iii) [22]. In the last case, as the hydroxy moiety is not a good leaving group, such alcohols were in situ converted into the corresponding O-allyl carbamates as leaving groups, followed by their reaction with imidazoles, affording the SN2’ products 3 (Scheme 1, reaction 1, iii).
![[1860-5397-19-93-i1]](/bjoc/content/inline/1860-5397-19-93-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of N-substituted imidazole derivatives from MBH adducts.
Scheme 1: Synthesis of N-substituted imidazole derivatives from MBH adducts.
Correlatively, we have previously reported a direct amination of cyclic MBH alcohols 4 with morpholine in the presence of imidazole (2a), as a powerful nucleophilic additive, affording, via competitive allylic nucleophilic substitution in toluene at reflux, a mixture of the corresponding N-substituted morpholine and N-substituted imidazole derivatives 6 [23]. In addition, a literature survey showed that nucleophilic allylic substitution reactions of acyclic/cyclic MBH adducts 1, 4, or 5, bearing good or poor leaving groups, using imidazole derivatives as nucleophilic reagents, have not been extensively developed. Therefore, in continuation of our previous study on the nucleophilic allylic substitution of MBH adducts [23-26], we disclose in this work a simple and efficient procedure for the synthesis of N-substituted imidazoles 6–8. The products were obtained either through direct conversions of the corresponding cyclic MBH alcohols 4 as well as acetates 5 in the presence of imidazoles 2a and 2b as nucleophilic reagents without catalysts or activating agents (Scheme 1, reaction 2), or from acyclic MBH alcohols 1 using DABCO as a powerful nucleophilic additive (Scheme 1, reaction 3).
Results and Discussion
In our first investigations, we selected the reaction of the primary acetate 5a [27] as the model substrate bearing a good leaving group, with imidazole (2a, 2 equiv) as a powerful nucleophilic reagent. The reaction was achieved with no need of a catalyst or any additive in toluene at reflux affording within 24 h the SN2-type product 6a in 82% yield (Table 1, entry 1). Similarly, the five-membered acetate 5b reacted under the same conditions and gave the N-allyl-substituted imidazole 6b in 65% yield (Table 1, entry 2).
Table 1: Allylation of imidazole derivatives 2a,b with cyclic MBH adducts 4 and 5.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-93-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | MBH adduct 4 or 5 |
Imidazole
2a or 2b |
Time (h) |
Product
6 or 7 |
Yield (%)
6 or 7 |
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-93-i4.svg?max-width=637&scale=1.0)
5a |
2a | 24 | 6a | 82 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-93-i5.svg?max-width=637&scale=1.0)
5b |
2a | 24 | 6b | 65 |
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-19-93-i6.svg?max-width=637&scale=1.0)
5c |
2a | 24 | 6c | 75 |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-19-93-i7.svg?max-width=637&scale=1.0)
5c |
2b | 48 | 7a | 87 |
| 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-19-93-i8.svg?max-width=637&scale=1.0)
5d |
2a | 24 | 6d | 69 |
| 6 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-19-93-i9.svg?max-width=637&scale=1.0)
4a |
2a | 48 | 6a | 88 |
| 7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-19-93-i10.svg?max-width=637&scale=1.0)
4b |
2a | 24 | 6b | 55 |
| 8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-19-93-i11.svg?max-width=637&scale=1.0)
4a |
2b | 72 | 7b | 80 |
| 9 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-19-93-i12.svg?max-width=637&scale=1.0)
4b |
2b | 72 | 7c | 72 |
| 10 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-19-93-i13.svg?max-width=637&scale=1.0)
4c |
2a | 48 | 6c | 76 |
| 11 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-19-93-i14.svg?max-width=637&scale=1.0)
4d |
2a | 24 | 6d | 60 |
| 12 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-19-93-i15.svg?max-width=637&scale=1.0)
4d |
2b | 72 | 7d | 85 |
Furthermore, treatment of the available secondary acetates 5c,d (R = Me) with imidazoles 2a and 2b (2 equiv) in refluxing toluene afforded the N-substituted imidazoles 6c, 6d, and 7a within ca. 24 h in 69–87% yields (Table 1, entries 3–5).
Having established the optimized conditions for the amination of primary and secondary acetates 5a–d (Table 1, entries 1–5), carrying a good leaving group (OAc), we turned our attention to the investigation of the direct amination of MBH alcohols 4a–d, with a poor leaving group (OH). Under the previous conditions (2 equiv of imidazole, toluene, reflux), the conversion of alcohol 4a [28] into the corresponding imidazole 6a was very slow and the starting materials were almost recovered. However, the continuous removal of water formed from the direct amination of alcohol 4a by azeotropic distillation shifted the position of the equilibrium in direction to the formation of the allylated imidazole 6a which was obtained in good 88% yield (Table 1, entry 6). The protocol was also successfully extended to the reaction of the primary five-membered alcohol 4b [29] with imidazole (2a) as well as to that of alcohols 4a,b with benzimidazole (2b), leading to the SN2-type products 6b and 7b,c, respectively, in 55–80% yields (Table 1, entries 7–9).
In addition, we have shown that the direct amination of the available secondary alcohols 4c,d (R = Me) [30] could be achieved with imidazole derivatives 2a,b under the conditions established above affording within 24–72 h the allylation products 6c,d and 7d in 60–85% yields (Table 1, entries 10–12).
Mechanistically, we believe that the nucleophilic allylic substitutions of alcohols 4, such as 4a, starts with a conjugate addition of imidazole (2a) at the C-β position of the Michael acceptor 4a, followed by elimination of the hydroxy moiety, affording the intermediate I. Similarly, a further second β’-conjugate addition of imidazole (2a) to I might occur, followed by elimination of imidazole (2a) finally providing the allylated derivative 6a (Scheme 2) [24-26,31]. It is notable, that such reaction mechanism involving the intermediate I was previously explored by Smith [32] and supported by studies of Tamura [33].
![[1860-5397-19-93-i2]](/bjoc/content/inline/1860-5397-19-93-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the allylation of imidazole with alcohol 4a.
Scheme 2: Proposed mechanism for the allylation of imidazole with alcohol 4a.
Next, in order to explore the scope of the above process, we have also investigated the direct allylation of imidazole (2a) with acyclic MBH alcohol 1a. In our first experiment, this substrate did not react with imidazole (2a) in toluene at reflux within 24 h, with or without azeotropic distillation, and the starting materials were completely recovered (Table 2, entry 1). Moreover, the addition of additives to the previous reaction mixture, such as DMAP [24-26,31] or molecular sieves 4 Å, commonly used to mediate nucleophilic allylic substitutions, did not lead to a notable improvement of the reaction outcome (Table 2, entries 2 and 3). However, the use of DABCO, commonly used as a powerful catalyst or a nucleophilic additive in the reaction of acyclic MBH adducts with various nucleophiles [21,34-37], did not afford the SN2/SN2’ products but provided the 1,4-adduct 8a in 84% yield (Table 2, entry 4).
![[Graphic 14]](/bjoc/content/inline/1860-5397-19-93-i16.svg?max-width=637&scale=1.0)
Alternatively, we also investigated the reaction of alcohol 1a [38] and imidazole (2a, 2 equiv), without any catalyst or additive in refluxing methanol, a solvent commonly employed in the conversion of MBH adducts using a variety of amines [39,40]. Our study showed that the imidazole (2a) reacted with alcohol 1a, without any additive or in the presence of DABCO as additive, in a 1,4-fashion leading to the imidazole derivative 8a, within 10 h in 65–68% yield (Table 2, entries 5 and 6).
Therefore, in our further experiments on the imidazole-mediated conversion of acyclic MBH alcohols, toluene at reflux was retained as the solvent of choice for the reaction using DABCO as additive.
Next, the treatment of acrylate-derived alcohols 1b,c (1b, EWG = CO2Et, R = Ph; 1c, EWG = CO2Me, R = Me) [41], under the previously optimized conditions afforded the corresponding 1,4-adducts 8b,c in 70–75% yield (Table 3, entries 2 and 3), as 55:45 and 59:41 mixtures of inseparable diastereomers, respectively. The relative diastereomeric ratios (dr) were determined by means of 1H NMR based on the proton at the α-position of the EWG moiety (Table 3).
Table 3: Michael addition of imidazole (2a) onto acyclic MBH alcohols 1a–f.
![[Graphic 15]](/bjoc/content/inline/1860-5397-19-93-i17.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Product | R | EWG | Solvent | Time (h) | 8, yield (%), dra |
| 1 | 8a | H | CO2Et | toluene | 24 | 84, none |
| 2 | 8b | Ph | CO2Et | toluene | 24 | 75, 55:45 |
| 3 | 8c | Me | CO2Me | toluene | 24 | 70, 59:41 |
| 4 | 8d | H | COPh | MeOH | 10 | 70, none |
| 5 | 8e | H | COn-Pr | MeOH | 12 | 76, none |
| 6 | 8f | H | COC6H11 | MeOH | 12 | 73, none |
aDetermined by 1H NMR spectroscopy from the crude reaction mixture.
In order to explore the scope of this synthetic approach, we have studied the reaction of ketone-derived alcohols such as 1d (EWG = COPh, R = H), 1e (EWG = COn-Pr, R = H), 1f (EWG = COc-C6H11, R = H) [42], and imidazole under the established reaction conditions and we have observed that the conversion was complete but was not clean. However, in methanol at reflux a clean reaction took place providing the corresponding 1,4-adducts 8d–f in 70–76% yields (Table 3, entries 4–6).
Conclusion
We have successfully developed an efficient N-nucleophilic allylic substitution protocol of cyclic MBH alcohols 4 and acetates 5 with imidazoles in refluxing toluene. The new N-substituted imidazoles 6 and 7 were afforded in high purity and good yields. In toluene or methanol at reflux temperature, acyclic MBH alcohols reacted with imidazole in a 1,4-fashion, leading to the corresponding Michael adducts 8 in 70–84% yields.
Synthetic applications of such imidazole derivatives [22,43,44], as well as their biological evaluation [45-47] are underway in our laboratory.
Experimental
Typical procedure for the α-substitution of cyclic MBH adducts with imidazoles
A mixture of allyl acetate 5a (2 mmol, 0.33 g) or allyl alcohol 4a (2 mmol, 0.25 g) and imidazole (2a, 4 mmol, 0.27 g) in toluene (25 mL) was heated under reflux (for 5a) or in a Dean–Stark apparatus (for 4a). After completion (TLC), the reaction mixture was cooled, washed with brine, and dried. The toluene was removed and the residue was purified by column chromatography on silica gel (acetone/ether 8:2) to give the pure N-substituted imidazole 6a.
Typical procedure for the preparation of imidazole derivatives 8
A mixture of acyclic MBH alcohol 1 (1 mmol), imidazole (2a, 2 mmol) and DABCO (1 mmol), was stirred at reflux temperature of methanol or toluene. After completion of the reaction, the solvent was removed by rotary evaporation and CH2Cl2 (10 mL) was added. The mixture was washed with brine and dried. Finally, the solvent was removed and the residue was purified by column chromatography on silica gel, using acetone/ether as eluent, to give the pure imidazole derivative 8.
Acknowledgements
We thank the Tunisian Chemical Society for the Eighteenth National Days of Chemistry JNC 2014 (see page 397 of http://docplayer.fr/57616885-Eighteenthnational-days-of-chemistry-organized-by-the-el-mouradi-skanes-hotel-skanesmonastir-tunisia.html).
References
-
Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–892. doi:10.1021/cr010043d
Return to citation in text: [1] [2] -
Basavaiah, D.; Reddy, B. S.; Badsara, S. S. Chem. Rev. 2010, 110, 5447–5674. doi:10.1021/cr900291g
Return to citation in text: [1] [2] -
Declerck, V.; Martinez, J.; Lamaty, F. Chem. Rev. 2009, 109, 1–48. doi:10.1021/cr068057c
Return to citation in text: [1] [2] -
Zhang, X.; Rao, W.; Sally; Chan, P. W. H. Org. Biomol. Chem. 2009, 7, 4186. doi:10.1039/b908447a
Return to citation in text: [1] [2] -
Kim, H. S.; Gowrisankar, S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2008, 49, 3858–3861. doi:10.1016/j.tetlet.2008.04.080
Return to citation in text: [1] [2] -
Kamimura, A.; Morita, R.; Matsuura, K.; Omata, Y.; Shirai, M. Tetrahedron Lett. 2002, 43, 6189–6191. doi:10.1016/s0040-4039(02)01318-7
Return to citation in text: [1] [2] -
Lee, C. G.; Lee, K. Y.; Lee, S.; Kim, J. N. Tetrahedron 2005, 61, 1493–1499. doi:10.1016/j.tet.2004.11.082
Return to citation in text: [1] [2] -
Gilchrist, T. L. Heterocyclic Chemistry, 3rd ed.; Longman: Harlow, UK, 1997; chapter 8.
Return to citation in text: [1] -
Botta, M.; Corelli, F.; Gasparrini, F.; Messina, F.; Mugnaini, C. J. Org. Chem. 2000, 65, 4736–4739. doi:10.1021/jo991937p
Return to citation in text: [1] -
Corelli, F.; Summa, V.; Brogi, A.; Monteagudo, E.; Botta, M. J. Org. Chem. 1995, 60, 2008–2015. doi:10.1021/jo00112a023
Return to citation in text: [1] -
Mhasni, O.; Elleuch, H.; Rezgui, F. Tetrahedron 2022, 111, 132732. doi:10.1016/j.tet.2022.132732
Return to citation in text: [1] -
Jana, U.; Maiti, S.; Biswas, S. Tetrahedron Lett. 2008, 49, 858–862. doi:10.1016/j.tetlet.2007.11.176
Return to citation in text: [1] -
Lee, K.-Y.; Lee, H.-S.; Kim, J.-N. Bull. Korean Chem. Soc. 2008, 29, 1099–1100. doi:10.5012/bkcs.2008.29.6.1099
Return to citation in text: [1] -
Liu, Y.-L.; Liu, L.; Wang, D.; Chen, Y.-J. Tetrahedron 2009, 65, 3473–3479. doi:10.1016/j.tet.2009.02.048
Return to citation in text: [1] -
Reddy, C. R.; Madhavi, P. P.; Reddy, A. S. Tetrahedron Lett. 2007, 48, 7169–7172. doi:10.1016/j.tetlet.2007.07.204
Return to citation in text: [1] -
Guo, S.; Song, F.; Liu, Y. Synlett 2007, 964–968. doi:10.1055/s-2007-973865
Return to citation in text: [1] -
Wu, W.; Rao, W.; Er, Y. Q.; Loh, J. K.; Poh, C. Y.; Chan, P. W. H. Tetrahedron Lett. 2008, 49, 2620–2624. doi:10.1016/j.tetlet.2008.02.079
Return to citation in text: [1] -
Yang, S.-C.; Chung, W.-H. Tetrahedron Lett. 1999, 40, 953–956. doi:10.1016/s0040-4039(98)02456-3
Return to citation in text: [1] -
Yang, S.-C.; Hsu, Y.-C.; Gan, K.-H. Tetrahedron 2006, 62, 3949–3958. doi:10.1016/j.tet.2006.02.035
Return to citation in text: [1] -
Ye, D.; Li, J.; Li, C.; Jia, X. Chin. J. Chem. 2009, 27, 1159–1162. doi:10.1002/cjoc.200990194
Return to citation in text: [1] -
Li, J.; Wang, X.; Zhang, Y. Tetrahedron Lett. 2005, 46, 5233–5237. doi:10.1016/j.tetlet.2005.05.107
Return to citation in text: [1] [2] -
Rodrigues, M. T., Jr.; Santos, M. S.; Santos, H.; Coelho, F. Tetrahedron Lett. 2014, 55, 180–183. doi:10.1016/j.tetlet.2013.10.146
Return to citation in text: [1] [2] -
Oueslati, Y.; Baioui, N.; Rezgui, F. Synth. Commun. 2017, 47, 892–898. doi:10.1080/00397911.2017.1295081
Return to citation in text: [1] [2] -
Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053
Return to citation in text: [1] [2] [3] -
Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106
Return to citation in text: [1] [2] [3] -
Ayadi, M.; Elleuch, H.; Vrancken, E.; Rezgui, F. Beilstein J. Org. Chem. 2016, 12, 2906–2915. doi:10.3762/bjoc.12.290
Return to citation in text: [1] [2] [3] -
Rezgui, F.; El Gaı̈ed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/s0040-4020(97)10023-0
Return to citation in text: [1] -
Rezgui, F.; El Gaied, M. M. Tetrahedron Lett. 1998, 39, 5965–5966. doi:10.1016/s0040-4039(98)01206-4
Return to citation in text: [1] -
Gatri, R.; El Gaı̈ed, M. M. Tetrahedron Lett. 2002, 43, 7835–7836. doi:10.1016/s0040-4039(02)01515-0
Return to citation in text: [1] -
Luo, S.; Wang, P. G.; Cheng, J.-P. J. Org. Chem. 2004, 69, 555–558. doi:10.1021/jo035345p
Return to citation in text: [1] -
Harrath, K.; Essalah, K.; Morell, C.; Chermette, H.; Boughdiri, S. Theor. Chem. Acc. 2015, 134, 98. doi:10.1007/s00214-015-1694-7
Return to citation in text: [1] [2] -
Smith, A. B., III; Wexler, B. A.; Slade, J. S. Tetrahedron Lett. 1980, 21, 3237–3240. doi:10.1016/s0040-4039(00)78655-2
Return to citation in text: [1] -
Tamura, R.; Tamai, S.; Katayama, H.; Suzuki, H. Tetrahedron Lett. 1989, 30, 3685–3688. doi:10.1016/s0040-4039(01)80483-4
Return to citation in text: [1] -
Chung, Y. M.; Gong, J. H.; Kim, T. H.; Kim, J. N. Tetrahedron Lett. 2001, 42, 9023–9026. doi:10.1016/s0040-4039(01)01971-2
Return to citation in text: [1] -
Lee, C.-G.; Gowrisankar, S.; Kim, J.-N. Bull. Korean Chem. Soc. 2005, 26, 481–484. doi:10.5012/bkcs.2005.26.3.481
Return to citation in text: [1] -
Ge, S.-Q.; Hua, Y.-Y.; Xia, M. Ultrason. Sonochem. 2009, 16, 743–746. doi:10.1016/j.ultsonch.2009.02.006
Return to citation in text: [1] -
Baidya, M.; Brotzel, F.; Mayr, H. Org. Biomol. Chem. 2010, 8, 1929. doi:10.1039/c000965b
Return to citation in text: [1] -
Villieras, J.; Rambaud, M. Org. Synth. 1988, 66, 220. doi:10.15227/orgsyn.066.0220
Return to citation in text: [1] -
Amri, H.; El Gaied, M. M.; Ben Ayed, T.; Villieras, J. Tetrahedron Lett. 1992, 33, 6159–6160. doi:10.1016/s0040-4039(00)60031-x
Return to citation in text: [1] -
Ben Ayed, T.; Amri, H.; El Gaïed, M. M.; Villiéras, J. Tetrahedron 1995, 51, 9633–9642. doi:10.1016/0040-4020(95)00538-j
Return to citation in text: [1] -
Yu, C.; Liu, B.; Hu, L. J. Org. Chem. 2001, 66, 5413–5418. doi:10.1021/jo015628m
Return to citation in text: [1] -
Ben Kraïem, J.; Ben Ayed, T.; Amri, H. Tetrahedron Lett. 2006, 47, 7077–7079. doi:10.1016/j.tetlet.2006.07.093
Return to citation in text: [1] -
Kobayashi, Y.; Kiyotsuka, Y.; Sugihara, Y.; Wada, K. Tetrahedron 2015, 71, 6481–6487. doi:10.1016/j.tet.2015.05.004
Return to citation in text: [1] -
Bellina, F.; Lessi, M.; Marianetti, G.; Panattoni, A. Tetrahedron Lett. 2015, 56, 3855–3857. doi:10.1016/j.tetlet.2015.04.094
Return to citation in text: [1] -
Devi, N.; Rawal, R. K.; Singh, V. Tetrahedron 2015, 71, 183–232. doi:10.1016/j.tet.2014.10.032
Return to citation in text: [1] -
Bellina, F.; Guazzelli, N.; Lessi, M.; Manzini, C. Tetrahedron 2015, 71, 2298–2305. doi:10.1016/j.tet.2015.02.024
Return to citation in text: [1] -
Aruchamy, B.; Drago, C.; Russo, V.; Pitari, G. M.; Ramani, P.; Aneesh, T. P.; Benny, S.; Vishnu, V. R. Eur. J. Pharm. Sci. 2023, 180, 106323. doi:10.1016/j.ejps.2022.106323
Return to citation in text: [1]
| 39. | Amri, H.; El Gaied, M. M.; Ben Ayed, T.; Villieras, J. Tetrahedron Lett. 1992, 33, 6159–6160. doi:10.1016/s0040-4039(00)60031-x |
| 40. | Ben Ayed, T.; Amri, H.; El Gaïed, M. M.; Villiéras, J. Tetrahedron 1995, 51, 9633–9642. doi:10.1016/0040-4020(95)00538-j |
| 41. | Yu, C.; Liu, B.; Hu, L. J. Org. Chem. 2001, 66, 5413–5418. doi:10.1021/jo015628m |
| 42. | Ben Kraïem, J.; Ben Ayed, T.; Amri, H. Tetrahedron Lett. 2006, 47, 7077–7079. doi:10.1016/j.tetlet.2006.07.093 |
| 1. | Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–892. doi:10.1021/cr010043d |
| 2. | Basavaiah, D.; Reddy, B. S.; Badsara, S. S. Chem. Rev. 2010, 110, 5447–5674. doi:10.1021/cr900291g |
| 3. | Declerck, V.; Martinez, J.; Lamaty, F. Chem. Rev. 2009, 109, 1–48. doi:10.1021/cr068057c |
| 9. | Botta, M.; Corelli, F.; Gasparrini, F.; Messina, F.; Mugnaini, C. J. Org. Chem. 2000, 65, 4736–4739. doi:10.1021/jo991937p |
| 10. | Corelli, F.; Summa, V.; Brogi, A.; Monteagudo, E.; Botta, M. J. Org. Chem. 1995, 60, 2008–2015. doi:10.1021/jo00112a023 |
| 22. | Rodrigues, M. T., Jr.; Santos, M. S.; Santos, H.; Coelho, F. Tetrahedron Lett. 2014, 55, 180–183. doi:10.1016/j.tetlet.2013.10.146 |
| 8. | Gilchrist, T. L. Heterocyclic Chemistry, 3rd ed.; Longman: Harlow, UK, 1997; chapter 8. |
| 23. | Oueslati, Y.; Baioui, N.; Rezgui, F. Synth. Commun. 2017, 47, 892–898. doi:10.1080/00397911.2017.1295081 |
| 1. | Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–892. doi:10.1021/cr010043d |
| 2. | Basavaiah, D.; Reddy, B. S.; Badsara, S. S. Chem. Rev. 2010, 110, 5447–5674. doi:10.1021/cr900291g |
| 3. | Declerck, V.; Martinez, J.; Lamaty, F. Chem. Rev. 2009, 109, 1–48. doi:10.1021/cr068057c |
| 4. | Zhang, X.; Rao, W.; Sally; Chan, P. W. H. Org. Biomol. Chem. 2009, 7, 4186. doi:10.1039/b908447a |
| 5. | Kim, H. S.; Gowrisankar, S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2008, 49, 3858–3861. doi:10.1016/j.tetlet.2008.04.080 |
| 6. | Kamimura, A.; Morita, R.; Matsuura, K.; Omata, Y.; Shirai, M. Tetrahedron Lett. 2002, 43, 6189–6191. doi:10.1016/s0040-4039(02)01318-7 |
| 7. | Lee, C. G.; Lee, K. Y.; Lee, S.; Kim, J. N. Tetrahedron 2005, 61, 1493–1499. doi:10.1016/j.tet.2004.11.082 |
| 20. | Ye, D.; Li, J.; Li, C.; Jia, X. Chin. J. Chem. 2009, 27, 1159–1162. doi:10.1002/cjoc.200990194 |
| 4. | Zhang, X.; Rao, W.; Sally; Chan, P. W. H. Org. Biomol. Chem. 2009, 7, 4186. doi:10.1039/b908447a |
| 5. | Kim, H. S.; Gowrisankar, S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2008, 49, 3858–3861. doi:10.1016/j.tetlet.2008.04.080 |
| 6. | Kamimura, A.; Morita, R.; Matsuura, K.; Omata, Y.; Shirai, M. Tetrahedron Lett. 2002, 43, 6189–6191. doi:10.1016/s0040-4039(02)01318-7 |
| 7. | Lee, C. G.; Lee, K. Y.; Lee, S.; Kim, J. N. Tetrahedron 2005, 61, 1493–1499. doi:10.1016/j.tet.2004.11.082 |
| 21. | Li, J.; Wang, X.; Zhang, Y. Tetrahedron Lett. 2005, 46, 5233–5237. doi:10.1016/j.tetlet.2005.05.107 |
| 15. | Reddy, C. R.; Madhavi, P. P.; Reddy, A. S. Tetrahedron Lett. 2007, 48, 7169–7172. doi:10.1016/j.tetlet.2007.07.204 |
| 17. | Wu, W.; Rao, W.; Er, Y. Q.; Loh, J. K.; Poh, C. Y.; Chan, P. W. H. Tetrahedron Lett. 2008, 49, 2620–2624. doi:10.1016/j.tetlet.2008.02.079 |
| 14. | Liu, Y.-L.; Liu, L.; Wang, D.; Chen, Y.-J. Tetrahedron 2009, 65, 3473–3479. doi:10.1016/j.tet.2009.02.048 |
| 18. | Yang, S.-C.; Chung, W.-H. Tetrahedron Lett. 1999, 40, 953–956. doi:10.1016/s0040-4039(98)02456-3 |
| 19. | Yang, S.-C.; Hsu, Y.-C.; Gan, K.-H. Tetrahedron 2006, 62, 3949–3958. doi:10.1016/j.tet.2006.02.035 |
| 12. | Jana, U.; Maiti, S.; Biswas, S. Tetrahedron Lett. 2008, 49, 858–862. doi:10.1016/j.tetlet.2007.11.176 |
| 13. | Lee, K.-Y.; Lee, H.-S.; Kim, J.-N. Bull. Korean Chem. Soc. 2008, 29, 1099–1100. doi:10.5012/bkcs.2008.29.6.1099 |
| 22. | Rodrigues, M. T., Jr.; Santos, M. S.; Santos, H.; Coelho, F. Tetrahedron Lett. 2014, 55, 180–183. doi:10.1016/j.tetlet.2013.10.146 |
| 43. | Kobayashi, Y.; Kiyotsuka, Y.; Sugihara, Y.; Wada, K. Tetrahedron 2015, 71, 6481–6487. doi:10.1016/j.tet.2015.05.004 |
| 44. | Bellina, F.; Lessi, M.; Marianetti, G.; Panattoni, A. Tetrahedron Lett. 2015, 56, 3855–3857. doi:10.1016/j.tetlet.2015.04.094 |
| 11. | Mhasni, O.; Elleuch, H.; Rezgui, F. Tetrahedron 2022, 111, 132732. doi:10.1016/j.tet.2022.132732 |
| 45. | Devi, N.; Rawal, R. K.; Singh, V. Tetrahedron 2015, 71, 183–232. doi:10.1016/j.tet.2014.10.032 |
| 46. | Bellina, F.; Guazzelli, N.; Lessi, M.; Manzini, C. Tetrahedron 2015, 71, 2298–2305. doi:10.1016/j.tet.2015.02.024 |
| 47. | Aruchamy, B.; Drago, C.; Russo, V.; Pitari, G. M.; Ramani, P.; Aneesh, T. P.; Benny, S.; Vishnu, V. R. Eur. J. Pharm. Sci. 2023, 180, 106323. doi:10.1016/j.ejps.2022.106323 |
| 28. | Rezgui, F.; El Gaied, M. M. Tetrahedron Lett. 1998, 39, 5965–5966. doi:10.1016/s0040-4039(98)01206-4 |
| 23. | Oueslati, Y.; Baioui, N.; Rezgui, F. Synth. Commun. 2017, 47, 892–898. doi:10.1080/00397911.2017.1295081 |
| 24. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 25. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 26. | Ayadi, M.; Elleuch, H.; Vrancken, E.; Rezgui, F. Beilstein J. Org. Chem. 2016, 12, 2906–2915. doi:10.3762/bjoc.12.290 |
| 27. | Rezgui, F.; El Gaı̈ed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/s0040-4020(97)10023-0 |
| 21. | Li, J.; Wang, X.; Zhang, Y. Tetrahedron Lett. 2005, 46, 5233–5237. doi:10.1016/j.tetlet.2005.05.107 |
| 34. | Chung, Y. M.; Gong, J. H.; Kim, T. H.; Kim, J. N. Tetrahedron Lett. 2001, 42, 9023–9026. doi:10.1016/s0040-4039(01)01971-2 |
| 35. | Lee, C.-G.; Gowrisankar, S.; Kim, J.-N. Bull. Korean Chem. Soc. 2005, 26, 481–484. doi:10.5012/bkcs.2005.26.3.481 |
| 36. | Ge, S.-Q.; Hua, Y.-Y.; Xia, M. Ultrason. Sonochem. 2009, 16, 743–746. doi:10.1016/j.ultsonch.2009.02.006 |
| 37. | Baidya, M.; Brotzel, F.; Mayr, H. Org. Biomol. Chem. 2010, 8, 1929. doi:10.1039/c000965b |
| 38. | Villieras, J.; Rambaud, M. Org. Synth. 1988, 66, 220. doi:10.15227/orgsyn.066.0220 |
| 33. | Tamura, R.; Tamai, S.; Katayama, H.; Suzuki, H. Tetrahedron Lett. 1989, 30, 3685–3688. doi:10.1016/s0040-4039(01)80483-4 |
| 24. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 25. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 26. | Ayadi, M.; Elleuch, H.; Vrancken, E.; Rezgui, F. Beilstein J. Org. Chem. 2016, 12, 2906–2915. doi:10.3762/bjoc.12.290 |
| 31. | Harrath, K.; Essalah, K.; Morell, C.; Chermette, H.; Boughdiri, S. Theor. Chem. Acc. 2015, 134, 98. doi:10.1007/s00214-015-1694-7 |
| 24. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 25. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 26. | Ayadi, M.; Elleuch, H.; Vrancken, E.; Rezgui, F. Beilstein J. Org. Chem. 2016, 12, 2906–2915. doi:10.3762/bjoc.12.290 |
| 31. | Harrath, K.; Essalah, K.; Morell, C.; Chermette, H.; Boughdiri, S. Theor. Chem. Acc. 2015, 134, 98. doi:10.1007/s00214-015-1694-7 |
| 32. | Smith, A. B., III; Wexler, B. A.; Slade, J. S. Tetrahedron Lett. 1980, 21, 3237–3240. doi:10.1016/s0040-4039(00)78655-2 |
| 29. | Gatri, R.; El Gaı̈ed, M. M. Tetrahedron Lett. 2002, 43, 7835–7836. doi:10.1016/s0040-4039(02)01515-0 |
| 30. | Luo, S.; Wang, P. G.; Cheng, J.-P. J. Org. Chem. 2004, 69, 555–558. doi:10.1021/jo035345p |
© 2023 Mhasni et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.