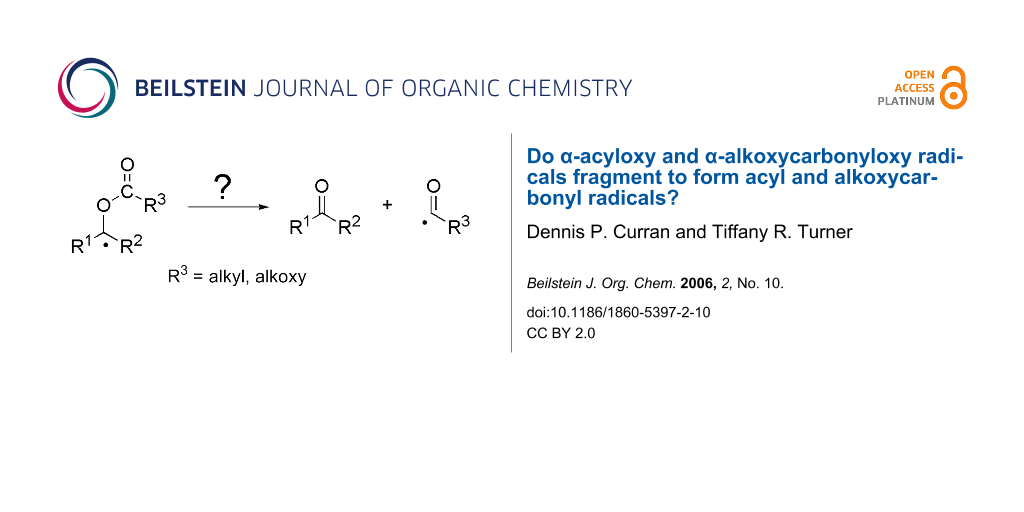
Abstract
The generation of α-acyloxy and α-alkoxycarbonyloxy radicals under reductive conditions in fragmentable probe experiments does not provide unequivocal evidence for the fragmentation of such radicals to give ketones and acyl or alkoxycarbonyl radicals. Instead, standard reduction predominates, even at low tin hydride concentrations. Some ketone product is formed in the α-acyloxy substrate at low concentrations, but it is unclear whether this product arises through a slow radical fragmentation process or an inefficient, chain-breaking oxidative process.

Graphical Abstract
Introduction
Most organic radical reactions occur through a cascade of two or more individual steps [1,2]. Knowledge of the nature and rates of these steps – in other words, the mechanism of the reaction – is of fundamental interest and is also important in synthetic planning. In synthesis, both the generation of the initial radical of the cascade and the removal of the final radical are crucial events [3]. Many useful radical reactions occur through chains that provide a naturally coupled regulation of radical generation and removal. Among the non-chain methods, generation and removal of radicals by oxidation and reduction are important, as is the "persistent radical effect" [4].
Recently, Wille and coworkers have described a collection of innovative new transformations that they have classed as "self-terminating radical reactions" [5-10]. For example, addition of broad assortment of oxygen-centered radicals to cyclodecyne 1 provides isomeric ketones 2 (major) and 3 (minor) in variable yields, depending on the specific radical involved and the reaction conditions. Representative reagents, reactions conditions and product yields for this very general transformation are shown in Figure 1.
![[1860-5397-2-10-1]](/bjoc/content/figures/1860-5397-2-10-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative self-terminating radical reactions.
Figure 1: Representative self-terminating radical reactions.
The suggested mechanism for formation of 2 involves addition of an oxygen-centered radical (XO•) to 1 to generate vinyl radical 4, followed by rapid radical translocation by 1,5-hydrogen atom transfer (Figure 2). The resulting radical 5 rebounds back to the enol ether in a 1,5-cyclization to provide 6. In the crucial self-terminating step, radical 6 is suggested to fragment to product 2 and radical X•. Related steps are involved in the formation of ketone 3 (not shown), except that the radical translocation occurs by 1,6-hydrogen transfer and the rebound cyclization is 1,6.
![[1860-5397-2-10-2]](/bjoc/content/figures/1860-5397-2-10-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Self-terminating, oxidative and chain mechanisms for evolution of 6 to 2.
Figure 2: Self-terminating, oxidative and chain mechanisms for evolution of 6 to 2.
The cascade in Figure 2 is a self-terminating, non-chain process if the radical X• does not continue on to propagate a chain in some way. Stable radicals such as X = NO2• or SO3•- and others are not expected to continue chain propagation. However, other radicals such as X = H•, alkyl (R•), acyl (RCO•) and alkoxycarbonyl (ROCO•) are quite reactive and might be expected to propagate chains under some conditions. Likewise, the stability of radicals X• is also important in the prior β-fragmentation step. If X• is a stable radical such as stannyl, benzyl or tert-alkyl, [11-15] then the fragmentation is well precedented [16]. However, for the hydrogen atom and carbon-centered radicals such as methyl and primary alkyl, the fragmentation has little precedent. Recent high level calculations support the notion that related fragmentations to make methyl radicals have high barriers and could be difficult to observe experimentally [17].
Because of the potential difficulties in β-fragmentation of some radicals X•, other pathways for product formation from 6 should be considered. Oxidation (6 → 7 → 2) is a relatively common pathway for electron rich radicals like 6 and can even occur under reducing conditions [18,19]. Cation 7 could evolve to ketone 2 by direct loss of X+ or through addition of a nucleophile (water or an alcohol, depending on conditions) to give an acetal-type intermediate that would in turn be subject to hydrolysis. In the case of thiohydroxamate precursors, radical 6 could also add back to the initial precursor in the standard Barton "group transfer" mechanism [20]. This would be followed by fragmentation to produce 8 (an acetal form of 2) and the starting radical XO•. This step begins a new propagation cycle in a chain.
We were especially interested in the general β-fragmentation reactions of radicals like 6 to provide either acyl or alkoxycarbonyl radicals (Figure 3). These radicals have many uses in synthesis, [21] so their generation by fragmentation could be a powerful tool. Because acyl and alkoxycarbonyl radicals are stabilized, it is not unreasonable to suggest that such a fragmentation could occur, yet there is nonetheless very little precedent [22,23].
![[1860-5397-2-10-3]](/bjoc/content/figures/1860-5397-2-10-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Proposed β-fragmentation reactions to form acyl and alkoxycarbonyl radicals.
Figure 3: Proposed β-fragmentation reactions to form acyl and alkoxycarbonyl radicals.
Are the fragmentation reactions in Figure 3 possible, and if so, then how fast are they? If not, then how does Wille's reaction work in such cases? To address these questions, we divorced the other steps of the Wille cascade to isolate the fragmentation reaction for a standard competition kinetic study [24,25]. The results of this study suggest that such fragmentations are very slow reactions at best. In turn, this leads us to suggest that some radicals in the Wille cascade progress to products by oxidation or group transfer rather than β-fragmentation.
Results and Discussion
We choose to generate the candidate radicals for fragmentation by a radical cyclization rather than by a standard atom or group abstraction reaction because the precursors are readily available and stable (α-halo acetates and carbonates are not stable) and because the intermediate radicals resemble Wille's typical intermediates. The syntheses of precursors 11a and 11b are summarized in Scheme 1.
![[1860-5397-2-10-i1]](/bjoc/content/inline/1860-5397-2-10-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of fragmentation probe substrates 11a,b
Scheme 1: Synthesis of fragmentation probe substrates 11a,b
Copper-mediated conjugate addition of 3-butenylmagnesium bromide to 3-methylcyclohexenone followed by quenching with either acetyl chloride [26] or methyl chloroformate provided enol ester 9a (50%) and enol carbonate 9b (83%). Oxidative cleavage [27-29] and reduction then provided alcohols 10a and 10b, which were converted to iodides 11a and 11b through mesylates by a standard procedure. Iodides 11a and 11b were stable to heating at 120°C in C6D6 for 24 h, so polar pathways for product formation are not likely in the cyclization experiments described below.
The projected mechanism for cyclizations of 11a,b with Bu3SnH in a competition kinetics setting is illustrated in Figure 4. Abstraction of iodine from 11 produces alkyl radical 12, which will rapidly cyclize to give key intermediate α-acyloxy radical 13a or α-alkoxycarbonyloxy radical 13b. Partitioning of 13a,b between bimolecular reduction to give 14a,b and unimolecular fragmentation to give 15 and 16a,b is the competition step, and a standard plot of the ratio of products as a function of tin hydride concentration should provide a straight line passing through the origin if radical fragmentation competes with reduction. Alternatively, oxidation of 13a,b to cation 17a,b will ultimately also result in the formation of 15, but the concentration dependence of this process is not clear since the oxidation step is not fully understood.
![[1860-5397-2-10-4]](/bjoc/content/figures/1860-5397-2-10-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Competing mechanistic pathways for reaction of 11 with Bu3SnH.
Figure 4: Competing mechanistic pathways for reaction of 11 with Bu3SnH.
Based on the mechanism in Figure 4, authentic samples of all products expected from the cyclizations of 11a and 11b were synthesized as shown in Scheme 2. Copper-mediated conjugate addition of propyl magnesium bromide to 3-methylcyclohexenone followed by quenching with acetyl chloride or methyl chloroformate provided reduced, uncyclized products 18a,b. These products were not detected in any of the subsequent cyclization experiments. Preparative radical cyclization of enol ether 11a with tributyltin hydride (0.1 M) followed by chromatographic purification provided 14a in 95% yield as an inseparable 1:2 mixture of exo and endo isomers. Likewise, cyclization of enol ester 11b provided a 1:2 mixture of 14b-exo and 14b-endo in 68% isolated yield. The expected product of fragmentation for both substrates, ketone 15, is a known compound [30,31] that was prepared by reduction of acetate 14a to provide a mixture of stereoisomeric alcohols (50%), followed by Dess-Martin oxidation (50%) [32].
![[1860-5397-2-10-i2]](/bjoc/content/inline/1860-5397-2-10-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of authentic samples of products
Scheme 2: Synthesis of authentic samples of products
Competition kinetic reactions were conducted under standard conditions, as detailed in the Supporting Information. Briefly, stock solutions of the iodide 11a,b (1 equiv) and Bu3SnH (1.1 equiv) in C6H6 or C6D6 were diluted to the required concentration of tin hydride, then AIBN (0.2 equiv) and p-dimethoxybenzene (0.1–0.2 equiv, internal standard) were added. The resulting mixture was rapidly heated to reflux and the progress of the reaction was followed by GC until no further consumption of starting material was observed. Product yields and ratios were then determined by GC and 1H NMR analyses. The results of the two analyses were comparable (typically ± 5%), and only the GC results are shown in the Tables. The complete data set is contained in the Supporting Information.
The results of single experiments for the cyclization of enol carbonate 11b at 0.1 M, 0.01 M and 0.001 M are summarized in the upper part of Table 1 (entries 1–3). At the higher two concentrations, complete conversion of 11b was observed and reduced product 14b was formed in good yield. None of the directly reduced product 18b was observed even at the highest concentration, indicating that the intermediate radical cyclization is fast (kC > 106 s-1). Negligible amounts of ketone 15 (≤ 2%) were observed, and its yields were not dependent on the tin hydride concentration. Accordingly, no evidence was obtained for fragmentation of intermediate α-alkoxycarbonyloxy radical 13b. At the lowest tin hydride concentration (entry 3), the conversion stopped with 25% of the starting iodide remaining, but again only a trace of 15 (1%) was detected. These results suggest chain propagation problems at this concentration, which is near the dilution limit for typical radical chain reactions.
Table 1: Product Ratios in Bu3SnH Mediated Cyclizations of 11a,ba
![[Graphic 1]](/bjoc/content/inline/1860-5397-2-10-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Precursor | [Bu3SnH] | Yldb 14a,b | Yldb 15a,b | Recoveredb 13a,b | Total Yld |
|---|---|---|---|---|---|---|
| 1 | 11b c | 0.1 M | 80% | 2% | - | 82% |
| 2 | 11b c | 0.01 M | 70% | 1% | - | 71% |
| 3 | 11b c | 0.001 M | 60% | 1% | 25% | 86% |
| 4 | 11a d | 0.1 M | 95% | 2% | - | 97% |
| 5 | 11a d | 0.01 M | 73% | 8% | - | 81% |
| 6 | 11a d | 0.005 M | 28% | 16% | 42% | 86% |
| 7 | 11a d | 0.001 M | 1% | 16% | 43% | 60% |
a) C6H6 or C6D6, 80°C, b) GC yield against p-dimethoxybenzene standard; 2-3/1 mixture of stereoisomers, c) single experiment, d) average of three experiments.
The results for cyclization of enol acetate 11a at four different concentrations are shown in the lower part of Table 1 (entries 4–7). Since increased amounts of ketone 15 were detected, these reactions were conducted in triplicate, and Table 1 records the averages of the three runs. The raw data in the Supporting Information show satisfactory (± 5% or less) agreement from run to run.
At 0.1 M (entry 4), the reaction of 11a goes to complete conversion and provides a high yield of reduced product 14a (95%) along with a trace of ketone 15 (2%). At 0.01 (entry 5), the conversion is again complete and yields of 14a and 15 are now 73% and 8%, respectively. However, as the reaction is diluted to 0.005 M (entry 6), the conversion of 11a becomes incomplete (42% recovery), while the yield of 14a declines to 28% and that of ketone 15 increases to 16%. Finally, at 0.001 M (entry 7), the yield of recovered 11a is still substantial (43%), while the amount of ketone 15 has stayed the same (16%) and the amount of the cyclized product 14a dropped to only 1%. A significant amount (40%) of the initial mass balance is unaccounted for in the three experiments at this concentration.
At first glance, the appearance of significant amounts of ketone 15 in the experiments with 11a at lower concentrations seems to support the fragmentation of radical 13a to release an acyl radical 16a. However, the ratios of 15/14a do not fit well with the standard model of competing unimolecular (fragmentation) and bimolecular (reduction) reactions in Figure 4. For example, the 10-fold dilution in going from entry 4 to entry 5 should have resulted in a 15/14a ratio about two times higher then was observed. In contrast, the small change in concentration going from entry 6 to 7 now results in an inordinately large increase in this ratio.
We feel that the results in Table 1 with 11a might be better accommodated by an oxidation pathway for conversion of radical 13a to ketone 15 via cation 17a. Since the nature of the oxidant is not known, it is not possible to interpret the concentration dependence of the product ratios. However, the trends of decreased conversions, decreased yields and lost mass balance are not uncommon in such radical oxidation reactions, especially those run under ostensibly reducing conditions [9]. The oxidation step may be inefficient and is almost surely a chain-breaking event. Thus, when the rate of the unspecified oxidation reaction(s) begins to exceed the rate of reduction of radicals 13a by tin hydride, the whole process begins to break down, so low conversions and yields result.
AIBN has been suggested to be an oxidant in related reactions, [9,33] so we conducted a series of individual cyclizations of 11a at 0.01 M with increasing amounts of AIBN. The results of these experiments are summarized in Table 2. If AIBN is acting as an oxidant, then the yield of 14a should decrease and 15 should increase as the concentration of AIBN increases. These trends were not observed. Instead, the yield of 14a stayed about constant, while the yield of 15 decreased by a small amount. These experiments do not support the active role of AIBN as anything other than a standard radical chain initiator.
Conclusion
In summary, the results with fragmentation probes 11a and 11b show the β-fragmentation reactions of α-acyloxy and α-alkoxycarbonyloxy radicals to give ketones and acyl or alkoxycarbonyl radicals (Figure 3 and Figure 4) are, at best, slow reactions. Only traces of ketone 15 were detected in the reduction of 11b even at very low concentrations, and a conservative upper limit for the fragmentation of this type of radical at 80°C is <103 s-1. Small but variable amounts of ketone 15 (7–16%) were produced during cyclizations of 11b, so the related α-acyloxy radical fragmentations to give acyl radicals could have rate constants as high as 103 – 104 s-1. However, the results can also be interpreted through the intermediacy of cationic precursors of ketones produced by radical oxidation, in which case the rate constant for fragmentation is even smaller. Even if the β-fragmentation is occurring by a radical pathway, it is so slow as to have limited synthetic value in radical chain sequences. The sluggishness of these β-fragmentation reactions is surprising, especially give that they produce a strong C=O bond and a stable radical.
In the bigger picture, the results suggest that continued evaluation of the role of β-fragmentation reactions in self-terminating oxidative radical reactions is worthwhile. While the reaction conditions of our probe experiments and prior preparative experiments are very different, the slowness of the β-fragmentations to produce acyl and alkoxycarbonyl radicals suggests that such reactions may not be very competitive under any standard preparative conditions. If fragmentations do not occur to produce acyl and alkoxycarbonyl radicals with reasonable rate constants, then it is unlikely that fragmentations to produce unstable alkyl radicals (for example, CH3•) or a hydrogen atom (H•) will occur. A similar conclusion has recently been reached through calculations by Sigmund, Wille, and Schiesser [17]. Either oxidative processes or group transfer reactions may contribute ketone formation in many of these types of reactions.
Oxidative pathways should also be considered when inorganic radicals such as NO3• and SO4•- are used as promoters. In such cases, the radicals produced on β-fragmentation (NO2• and SO3•-) are very stable, so the proposed fragmentation is more likely. However, the inorganic conditions are also more strongly oxidizing. So both oxidation and fragmentation pathways are seem reasonable, and further experimentation will be needed to identify which path is preferred as a function of reaction conditions and fragmenting radical in these cases.
The synthetic value of self-terminating oxidative radical reactions is already evident from the pioneering work of Wille, and added value will accrue as we continue to better understand the details of each of the different processes for conducting such reactions.
Supporting Information
| Supporting Information File 1: Complete experimental details and full spectroscopic data for all new compounds; procedures and data for individual competition kinetic experiments and control experiments (7 pages). | ||
| Format: DOC | Size: 119.5 KB | Download |
References
-
Zard, S. Z. Radical Reactions in Organic Synthesis; Oxford University Press: Oxford, 2003.
Return to citation in text: [1] -
Renaud, P.; Sibi, M. P. Radicals in Organic Synthesis, 1st ed.; Wiley-VCH: Weinheim, 2001; pp 1–2.
Return to citation in text: [1] -
Curran, D. P. Synlett 1991, 63–72. doi:10.1055/s-1991-20631
Return to citation in text: [1] -
Studer, A. Chem.–Eur. J. 2001, 7, 1159–1164. doi:10.1002/1521-3765(20010316)7:6<1159::AID-CHEM1159>3.0.CO;2-I
Return to citation in text: [1] -
Dreessen, T.; Jargstorff, C.; Lietzau, L.; Plath, C.; Stademann, A.; Wille, U. Molecules 2004, 9, 480–497.
review.
Return to citation in text: [1] -
Wille, U. Chem.–Eur. J. 2002, 8, 340–347. doi:10.1002/1521-3765(20020118)8:2<340::AID-CHEM340>3.0.CO;2-4
review.
Return to citation in text: [1] -
Wille, U.; Jargstorff, C. J. Chem. Soc., Perkin Trans. 1 2002, 1036–1041. doi:10.1039/b201672a
Return to citation in text: [1] -
Wille, U. Tetrahedron Lett. 2002, 43, 1239–1242. doi:10.1016/S0040-4039(01)02398-X
Return to citation in text: [1] -
Wille, U. J. Am. Chem. Soc. 2002, 124, 14–15. doi:10.1021/ja017006o
Return to citation in text: [1] [2] [3] -
Stademann, A.; Wille, U. Aust. J. Chem. 2004, 57, 1055–66. doi:10.1071/CH04124
Return to citation in text: [1] -
Roepel, M. G. Tetrahedron Lett. 2002, 43, 1973–1976. doi:10.1016/S0040-4039(02)00169-7
Return to citation in text: [1] -
Cai, Y. D.; Roberts, B. P. Tetrahedron Lett. 2003, 44, 4645–4648. doi:10.1016/S0040-4039(03)01092-X
Return to citation in text: [1] -
Cai, Y. D.; Dang, H. S.; Roberts, B. P. Tetrahedron Lett. 2004, 45, 4405–4409. doi:10.1016/j.tetlet.2004.03.165
Return to citation in text: [1] -
Cai, Y. D.; Roberts, B. P. Tetrahedron Lett. 2004, 45, 1485–1488. doi:10.1016/j.tetlet.2003.12.045
Return to citation in text: [1] -
Cai, Y. D.; Roberts, B. P.; Tocher, D. A.; Barnett, S. A. Org. Biomol. Chem. 2004, 2, 2517–2529. doi:10.1039/b407215b
Return to citation in text: [1] -
Rosenstein, I. In Radicals in Organic Synthesis, 1st ed.; Renard, P.; Sibi, M., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 1, pp 50–71.
review.
Return to citation in text: [1] -
Sigmund, D.; Schiesser, C. H.; Wille, U. Synthesis 2005, 1437–1444.
Return to citation in text: [1] [2] -
Beckwith, A. L. J.; Bowry, V. W.; Bowman, W. R.; Mann, E.; Parr, J.; Storey, J. M. D. Angew. Chem., Int. Ed. 2004, 43, 95–98. doi:10.1002/anie.200352419
Return to citation in text: [1] -
Bowman, W. R.; Heaney, H.; Jordan, B. M. Tetrahedron 1991, 47, 10119–10128. doi:10.1016/S0040-4020(01)96061-2
Return to citation in text: [1] -
Motherwell, W. B.; Imboden, C. In Radicals in Organic Synthesis, 1st ed.; Renard, P.; Sibi, M., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 1, pp 109–130.
It also merits consideration that thiyl radicals present in such reactions could add to the alkyne and evolve to radicals like 6 with ArS in place of OX. These could potentially progress on to ketone products either by oxidation or group transfer.
Return to citation in text: [1] -
Chatgilialoglu, H.; Crich, D.; Komatsu, M.; Ryu, I. Chem. Rev. 1999, 99, 1991–2069. doi:10.1021/cr9601425
Return to citation in text: [1] -
Horman, I.; Freidrich, S.; Keefer, R. M.; Andrews, L. J. J. Org. Chem. 1969, 34, 905–911. doi:10.1021/jo01256a028
Fragmentations to form acyl radicals have been suggested in NBS brominations, but the products of these reactions can also be rationalized by standard ionic mechanisms, see also reference 23.
Return to citation in text: [1] -
Anson, M. S.; Montana, J. G. Synlett 1994, 219–220. doi:10.1055/s-1994-22802
Return to citation in text: [1] -
Newcomb, M. Tetrahedron 1993, 49, 1151–76. doi:10.1016/S0040-4020(01)85808-7
Return to citation in text: [1] -
Newcomb, M. In Radicals in Organic Synthesis, 1st ed.; Renard, P.; Sibi, M., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 1, pp 317–336.
Return to citation in text: [1] -
Silvestri, M. G.; Hanson, M. P.; Pavlovich, J. G.; Studen, L. F.; DeClue, M. S.; DeGraffenreid, M. R.; Amos, C. D. J. Org. Chem. 1999, 64, 6597–6602. doi:10.1021/jo9824807
Return to citation in text: [1] -
Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispino, G. A.; Hartung, J.; Jeong, S. K.; Kwong, H. L.; Morikawa, K.; Wang, Z. M. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003
Return to citation in text: [1] -
Kodama, M.; Shiobara, Y.; Sumimoto, H.; Mitani, K.; Ueno, K. Chem. Pharm. Bull. 1987, 35, 4039–4042.
Return to citation in text: [1] -
Areces, P.; Jimenez, J. L.; de la Cruz Pozo, M.; Roman, E.; Serrano, J. A. J. Chem. Soc., Perkin Trans. 1 2001, 754–762. doi:10.1039/b006078j
Return to citation in text: [1] -
Miyano, M.; Stealey, M. A. J. Org. Chem. 1982, 47, 3184–3186. doi:10.1021/jo00137a036
Return to citation in text: [1] -
Lo Cicero, B.; Weisbuch, F.; Dana, G. J. Org. Chem. 1981, 46, 914–919. doi:10.1021/jo00318a017
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070
Return to citation in text: [1] -
Curran, D. P.; Liu, H. T. J. Chem. Soc., Perkin Trans. 1 1994, 1377–1393. doi:10.1039/p19940001377
Return to citation in text: [1]
| 9. | Wille, U. J. Am. Chem. Soc. 2002, 124, 14–15. doi:10.1021/ja017006o |
| 33. | Curran, D. P.; Liu, H. T. J. Chem. Soc., Perkin Trans. 1 1994, 1377–1393. doi:10.1039/p19940001377 |
| 1. | Zard, S. Z. Radical Reactions in Organic Synthesis; Oxford University Press: Oxford, 2003. |
| 2. | Renaud, P.; Sibi, M. P. Radicals in Organic Synthesis, 1st ed.; Wiley-VCH: Weinheim, 2001; pp 1–2. |
| 11. | Roepel, M. G. Tetrahedron Lett. 2002, 43, 1973–1976. doi:10.1016/S0040-4039(02)00169-7 |
| 12. | Cai, Y. D.; Roberts, B. P. Tetrahedron Lett. 2003, 44, 4645–4648. doi:10.1016/S0040-4039(03)01092-X |
| 13. | Cai, Y. D.; Dang, H. S.; Roberts, B. P. Tetrahedron Lett. 2004, 45, 4405–4409. doi:10.1016/j.tetlet.2004.03.165 |
| 14. | Cai, Y. D.; Roberts, B. P. Tetrahedron Lett. 2004, 45, 1485–1488. doi:10.1016/j.tetlet.2003.12.045 |
| 15. | Cai, Y. D.; Roberts, B. P.; Tocher, D. A.; Barnett, S. A. Org. Biomol. Chem. 2004, 2, 2517–2529. doi:10.1039/b407215b |
| 30. | Miyano, M.; Stealey, M. A. J. Org. Chem. 1982, 47, 3184–3186. doi:10.1021/jo00137a036 |
| 31. | Lo Cicero, B.; Weisbuch, F.; Dana, G. J. Org. Chem. 1981, 46, 914–919. doi:10.1021/jo00318a017 |
| 5. |
Dreessen, T.; Jargstorff, C.; Lietzau, L.; Plath, C.; Stademann, A.; Wille, U. Molecules 2004, 9, 480–497.
review. |
| 6. |
Wille, U. Chem.–Eur. J. 2002, 8, 340–347. doi:10.1002/1521-3765(20020118)8:2<340::AID-CHEM340>3.0.CO;2-4
review. |
| 7. | Wille, U.; Jargstorff, C. J. Chem. Soc., Perkin Trans. 1 2002, 1036–1041. doi:10.1039/b201672a |
| 8. | Wille, U. Tetrahedron Lett. 2002, 43, 1239–1242. doi:10.1016/S0040-4039(01)02398-X |
| 9. | Wille, U. J. Am. Chem. Soc. 2002, 124, 14–15. doi:10.1021/ja017006o |
| 10. | Stademann, A.; Wille, U. Aust. J. Chem. 2004, 57, 1055–66. doi:10.1071/CH04124 |
| 32. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070 |
| 4. | Studer, A. Chem.–Eur. J. 2001, 7, 1159–1164. doi:10.1002/1521-3765(20010316)7:6<1159::AID-CHEM1159>3.0.CO;2-I |
| 26. | Silvestri, M. G.; Hanson, M. P.; Pavlovich, J. G.; Studen, L. F.; DeClue, M. S.; DeGraffenreid, M. R.; Amos, C. D. J. Org. Chem. 1999, 64, 6597–6602. doi:10.1021/jo9824807 |
| 27. | Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispino, G. A.; Hartung, J.; Jeong, S. K.; Kwong, H. L.; Morikawa, K.; Wang, Z. M. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003 |
| 28. | Kodama, M.; Shiobara, Y.; Sumimoto, H.; Mitani, K.; Ueno, K. Chem. Pharm. Bull. 1987, 35, 4039–4042. |
| 29. | Areces, P.; Jimenez, J. L.; de la Cruz Pozo, M.; Roman, E.; Serrano, J. A. J. Chem. Soc., Perkin Trans. 1 2001, 754–762. doi:10.1039/b006078j |
| 20. |
Motherwell, W. B.; Imboden, C. In Radicals in Organic Synthesis, 1st ed.; Renard, P.; Sibi, M., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 1, pp 109–130.
It also merits consideration that thiyl radicals present in such reactions could add to the alkyne and evolve to radicals like 6 with ArS in place of OX. These could potentially progress on to ketone products either by oxidation or group transfer. |
| 22. |
Horman, I.; Freidrich, S.; Keefer, R. M.; Andrews, L. J. J. Org. Chem. 1969, 34, 905–911. doi:10.1021/jo01256a028
Fragmentations to form acyl radicals have been suggested in NBS brominations, but the products of these reactions can also be rationalized by standard ionic mechanisms, see also reference 23. |
| 23. | Anson, M. S.; Montana, J. G. Synlett 1994, 219–220. doi:10.1055/s-1994-22802 |
| 18. | Beckwith, A. L. J.; Bowry, V. W.; Bowman, W. R.; Mann, E.; Parr, J.; Storey, J. M. D. Angew. Chem., Int. Ed. 2004, 43, 95–98. doi:10.1002/anie.200352419 |
| 19. | Bowman, W. R.; Heaney, H.; Jordan, B. M. Tetrahedron 1991, 47, 10119–10128. doi:10.1016/S0040-4020(01)96061-2 |
| 24. | Newcomb, M. Tetrahedron 1993, 49, 1151–76. doi:10.1016/S0040-4020(01)85808-7 |
| 25. | Newcomb, M. In Radicals in Organic Synthesis, 1st ed.; Renard, P.; Sibi, M., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 1, pp 317–336. |
| 16. |
Rosenstein, I. In Radicals in Organic Synthesis, 1st ed.; Renard, P.; Sibi, M., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 1, pp 50–71.
review. |
| 21. | Chatgilialoglu, H.; Crich, D.; Komatsu, M.; Ryu, I. Chem. Rev. 1999, 99, 1991–2069. doi:10.1021/cr9601425 |
© 2006 Curran and Turner; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)