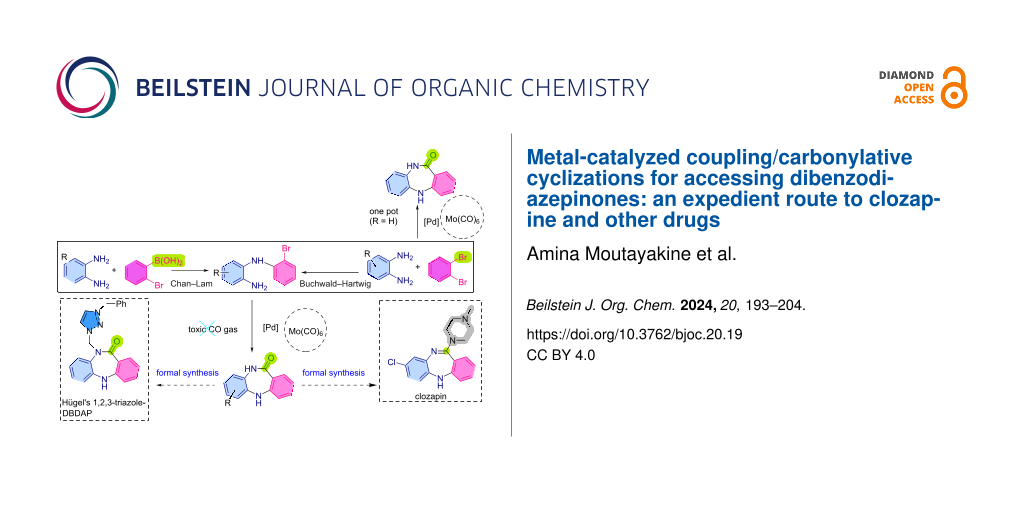
Abstract
A sequential strategy to access 10,11-dihydro-5H-dibenzo[b,e][1,4]diazepinones (DBDAPs) is disclosed in this article through a palladium and copper-catalyzed amination (Buchwald–Hartwig (B–H) or Chan–Lam (C–L)) followed by a palladium-catalyzed intramolecular aminocarbonylation with Mo(CO)6 as CO surrogate (to avoid toxic CO handling) of readily available o-phenylenediamines and either 1,2-dibromobenzene or 2-bromophenylboronic acid. The 10,11-dihydro-5H-dibenzo[b,e][1,4]diazepinone could be synthezised in good yield using a sequential catalytic procedure, using both C–L and B–H approaches. Gratifingly, the use of the C–L reaction was more impressive, and afforded the dibenzodiazepinones in good yields (up to 45%; 2 steps) and much milder conditions using copper as the catalyst. The synthetic utility of this novel strategy was showcased by demonstrating a formal synthesis for the antipsychotic drug clozapine and to an anticancer triazole–DBDAP hybrid.

Graphical Abstract
Introduction
Dibenzodiazepine units are without doubt highly privileged structures, endowed with numerous medicinally relevant properties, and notably include anti-anxiolytic and antidepressant activities. These scaffolds have received much interest from the medicinal chemist community, which led to the development of several antidepressant agents such as dibenzepin, sintamil, as well as the well-known medication, clozapine, an FDA-approved atypical antipsychotic drug, that has been adopted as a treatment of schizophrenia and schizoaffective disorders (Figure 1) [1,2]. Dibenzodiazepinones were also found to exhibit significant anticancer properties [3], as they were found to effectively inhibit tumor invasion in vitro [4], and induce apoptosis among several cancer cell lines [5]. Additionally, several dibenzodiazepinone-based structures were proven to act as p21-activated kinase (PAK) inhibitors [6], and Chk1 inhibitors [7]. The abovementioned pharmaceutical properties of the dibenzodiazepinone class have driven the development of novel synthetic strategies leading to these scaffolds in a step-economical and greener manner. Our previous review in 2018 focused on a variety of routes to these compounds [8].
![[1860-5397-20-19-1]](/bjoc/content/figures/1860-5397-20-19-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active dibenzodiazepinones.
Figure 1: Biologically active dibenzodiazepinones.
The well-known Buchwald–Hartwig (B–H) and Chan–Lam (C–L) reactions have proven to be highly useful procedures that allow the step-economical synthesis of diverse biologically relevant heterocycles through C–N bond formation [9]. These approaches resulted in shortening the synthetic routes that were widely employed to access these heterocyclic scaffolds. Over the last decades, the Chan–Lam coupling reaction has drawn great attention among the synthetic chemistry community which contributed to the development of various synthetic routes to relevant heterocycles in high efficiency [10]. The Chan–Lam coupling is considered a greener alternative to traditional C–N coupling reactions, as it can be carried out under mild reaction conditions (room temperature and short reaction times, etc.), plus it does not require expensive metals like Pd, being carried out with Cu.
The aminocarbonylation reaction which was introduced by Schoenberg and Heck in 1974 is an efficient catalytic route to carboxamides [11]. It was a major step forward and has been amply applied in a number of carbonylation reactions over the years [12].
In 2011, Buchwald and Tsvelikhovsky introduced an efficient synthetic strategy to construct diverse dibenzodiazepinones through a sequential methodology consisting of a B–H coupling between o-carbonylanilines and 1,2-dihaloarene derivatives providing access to key precursors that undergo a tandem amination–intramolecular cyclization via a cross-coupling reaction with NH3 [13]. The reaction was undertaken in the presence of a catalytic amount of a palladium catalyst and afforded a library of dibenzodiazepinones in good to excellent yields (Scheme 1a).
![[1860-5397-20-19-i1]](/bjoc/content/inline/1860-5397-20-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Different synthetic routes to DBDAPs (a–c), including our novel approach (d).
Scheme 1: Different synthetic routes to DBDAPs (a–c), including our novel approach (d).
In 2013, Zhang et al. developed a synthetic route leading to structurally diverse dibenzodiazepinones via a copper-catalyzed C–N bond coupling between 2-halobenzoates and o-phenylenediamines leading to a key intermediate that undergoes an intramolecular N-acylation to afford the corresponding dibenzodiazepinone structure in high yields (Scheme 1b) [14]. Another innovative strategy was reported by Laha et al., aiming to access dibenzodiazepinone structures via a double N-arylation of 2-aminobenzamides with 1,2-dihaloarenes using a palladium-based catalytic system [15].
Mechanistic investigations supported the fact that the regioselective N-arylation of the 2-aminobenzamide occurs first at the amide position. This approach enabled the successful synthesis of a broad spectrum of dibenzodiazepinone units in a one-pot fashion. The synthetic utility of Laha’s approach was highlighted by preparing the corresponding dibenzodiazepinone which was further reacted with N-methylpiperazine in the presence of TiCl4 to afford clozapine, an antipsychotic drug.
We now disclose a different, but facile approach to access several 10,11-dihydro-5H-dibenzo[b,e][1,4]diazepinones, using a sequential Chan–Lam and Buchwald–Hartwig intramolecular aminocarbonylation (Scheme 1d). This approach has not been reported previously (the methods a–c indicated in Scheme 1 have a different route, and none involve either a Chan–Lam or carbonylative cyclization). For the sake of health and safety, and given that our infrastructures did not permit the use of molecular CO, we felt more secure with a suitable surrogate. Not only that, the use of safer to use surrogates, is important for use in enabling technologies, like continuous flow and microwave-heated reactions. In fact, CO-free aminocarbonylation reactions are well known, and molybdenum hexacarbonyl (Mo(CO)6) is a very useful surrogate having been used in a multitude of reactions [16,17].
The present approach enables the formation of two C–N bonds along with a C–C bond and provides a good alternative to previously reported strategies, as it enables the formation of these structures in a multicomponent fashion in the presence of a CO surrogate through the in situ formation of an o-(2-bromophenyl)aminoaniline intermediate (Scheme 1d). It should be noted these target compounds have been of great interest to our group and in 2015 we reported a proposed novel methodology for the synthesis of dibenzodiazepines [18], however, upon later careful review of the product structure it was revealed that the purported dibenzodiazepine products were, in fact, diarylimines, which resulted from a nucleophilic addition of the aniline reagents to the aldimine substrates, followed by elimination of an tosylamine product. This was one of the principle driving forces for the development of the work discussed in this report.
Results and Discussion
Synthesis of o-(2-bromophenyl)aminoaniline via Buchwald–Hartwig C–N coupling
One-pot synthesis of dibenzodiazepinones
Our preliminary attempt to synthesize DBDAPs via B–H amination and carbonylation was carried out in the presence of o-phenylenediamine (1a) and 1,2-dibromobenzene (2) as model reactants using Pd(OAc)2 in combination with t-BuXPhos (2-di-tert-butylphosphino-2',4',6'-triisopropyl-1,1'-biphenyl) (5 mol %), and Et3N (2.5 equiv) as base in DMF. In this case, DMF served as the CO surrogate, as it was disclosed that DMF, the reaction solvent, could act as a potential carbon monoxide surrogate under certain circumstances, notably, in metal-catalyzed aminocarbonylation procedures [19,20]. Unfortunately, no DBDAP was obtained and we only observed the formation of intermediate 3a in 25% yield (entry 1, Table 1). Next, the same procedure was carried out in the presence of molybdenum hexacarbonyl (Mo(CO)6, 2 equiv) as CO surrogate, under the previous conditions, but again we only observed the formation of intermediate 3a in 21% yield (entry 2, Table 1). Changing the ligand to triphenylphosphine, did not provide any improvement of the reaction outcome as only traces of the intermediate 3a were obtained (entry 3, Table 1). Switching to DBU as the base under these conditions, gave intermediate 3a in 35% yield (entry 4, Table 1). In fact, DBU was previously shown by Wannberg and Larhed to be an effective ligand in a variety of highly efficient aminocarbonylation reactions with Mo(CO)6 due to its strong basicity and accelerated release of CO from this reagent [21]. The reaction was then screened using two different bidentate ligands, XPhos and XantPhos, and using the previous reaction conditions. However, we only obtained traces of intermediate 3a (entries 5 and 6, Table 1). A slight improvement of the yield of the intermediate 3 was obtained when using DBU in dioxane, which gave 3a in 42%, but only traces of the target DBDAP were observed (entry 7, Table 1). The difficulty encountered in the formation of DBDAP, prompted us to test alternative CO surrogates. When the reaction was performed using Co2(CO)8 (0.3 equiv) in the presence of DBU, the intermediate 3a was isolated in 35% yield, but again no DBDAP 4 was obtained (entry 8, Table 1). Formic acid, an effective CO surrogate [20,22], was also screened. The reaction was carried out in the presence of acetic anhydride (Ac2O) as an activator, but unfortunately, no DBDAP was obtained (entry 9, Table 1). The use of triphosgene as the CO transfer agent failed to give any product.
Table 1: Explorative study of the sequential Buchwald–Hartwig amination/Pd-catalyzed carbonylative cyclization leading to DBDAPs.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-19-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Cat (mol %) |
Ligand
(5 mol %) |
CO | Base | Solvent | 3ab | 4ab |
| 1 | Pd(OAc)2 (5) | t-BuXPhos | – | Et3N | DMF | 25% | 0 |
| 2 | Pd(OAc)2 (5) | t-BuXPhos | Mo(CO)6 | Et3N | DMF | 21% | 0 |
| 3 | Pd(OAc)2 (5) | PPh3 | Mo(CO)6 | Et3N | DMF | traces | 0 |
| 4 | Pd(OAc)2 (5) | t-BuXPhos | Mo(CO)6 | DBU | DMF | 35% | 0 |
| 5 | Pd(OAc)2 (5) | XPhos | Mo(CO)6 | Et3N | DMF | traces | 0 |
| 6 | Pd(OAc)2 (5) | XantPhos | Mo(CO)6 | Et3N | DMF | traces | 0 |
| 7c | Pd(OAc)2 (5) | t-BuXPhos | Mo(CO)6 | DBU | dioxane | 42% | traces |
| 8c | Pd(OAc)2 (5) | t-BuXPhos | Co2(CO)8 | DBU | DMF | 35% | – |
| 9c | Pd(OAc)2 (5) | t-BuXPhos | HCOOH/Ac2O | Et3N | DMF | – | – |
aReaction conditions: o-phenylenediamine (1a, 0.46 mmol), dibromobenzene (2, 0.46 mmol), Pd(OAc)2, base (2.5 equiv), CO surrogate (Mo(CO)6 and other CO surrogate (2 equiv) or Co2(CO)8 (0.3 equiv)), solvent (5 mL), 130 °C, 20 h. bIsolated yields. cThe reaction was carried out during 24 h.
When we undertook the B–H amination/carbonylative cyclization of o-phenylenediamine (1a) with 1,2-dibromobenzene (2) in the presence of 5 mol % of PdCl2(CH3CN)2 and 5 mol % of t-BuXPhos, with Et3N (2.5 equiv) and Mo(CO)6 in DMF at 150 °C, surprisingly this afforded the 5H-dibenzo[b,e][1,4]diazepin-11-ol (5), the tautomer of DBDAP (4a) in 53% (Scheme 2). We attempted to convert compound 5 into the keto form 4a by using TFA to shift the equilibrium towards the desired product, but this proved to be futile under these conditions, as only the iminol 5 structure was observed.
![[1860-5397-20-19-i2]](/bjoc/content/inline/1860-5397-20-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: One-pot synthesis of 5H-dibenzo[b,e][1,4]diazepin-11-ol (5).
Scheme 2: One-pot synthesis of 5H-dibenzo[b,e][1,4]diazepin-11-ol (5).
Attempt at accessing dibenzodiazepinone via step-wise synthesis
Due to the difficulty encountered in the one-step synthesis of DBDAPs, we embarked on an in-depth study of the B–H coupling/carbonylative cyclization in a step-wise fashion. Our first attempt was conducted using the previous conditions, which led to the desired compound 3a in 15% yield (entry 1, Table 2). Changing the ligand to PPh3 under the same conditions (entry 2, Table 2) resulted in poorer results, as only traces of the desired compound 3a were observed. Then, we considered XPhos (entry 3, Table 2), and the bidendate ligands XantPhos and DPEPhos (entries 4 and 5, Table 2), but no improvements were observed. Then, we tested an alternative palladium source, namely Pd2dba3, but again only traces of the compound 3a were observed (entry 6, Table 2). Next, we increased the Pd(OAc)2 catalyst loading to 10 mol % and the amount of the t-BuXphos ligand to 15 mol % in the presence of DBU and DMF. Under these conditions, the intermediate 3a was obtained in 15% yield along with the undesired dihydrophenazine 6 side product in 5% yield, produced by a further C–N bond coupling (entry 7, Table 2). We decided to decrease the amount of base and time, but little improvement was observed (entry 8, Table 2). Given the well-established role of the base on the B–H coupling, we considered exploring alternative bases. We conducted the reaction in the presence of the previously disclosed catalytic system but using t-BuOK as the base and obtained compound 3a in 22% yield (entry 9, Table 2). Conducting the reaction in the presence of Cs2CO3 in DMF, failed to provide any improvement (entry 10, Table 2). However, replacing DMF by dioxane as the solvent in the presence of DBU led to a significant improvement in the yield of the reaction, as the intermediate 3a could be obtained in 40% yield, along with the phenazine 6 in 48% (entry 11, Table 2). Next, we screened other ligands such as XPhos and DPEPhos in the presence of Cs2CO3 as base in dioxane, however, the undesired phenazine product 6 was still obtained in moderate yield under these conditions (entries 12 and 13, Table 2). In the presence of SPhos as ligand, Cs2CO3, and toluene as solvent, the desired intermediate 3a was obtained in 20% yield along with 55% of the phenazine 6 (entry 14, Table 2). Although toluene was shown to be a good solvent for this B–H coupling reaction, we were unable to prevent the double B–H reaction from occurring leading to the phenazine 6, even when shortening the reaction time to 1 h.
Table 2: Influence of the catalytic system, base, and solvent combination on the outcome of the Buchwald–Hartwig reaction.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-19-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Cat (mol %) | Ligand | Base | Solvent | Time | 3ab | 6b |
| 1 | Pd(OAc)2 (5) | t-BuXPhos | Et3N | DMF | 10 h | 15 | – |
| 2 | Pd(OAc)2 (5) | PPh3 | Et3N | DMF | 10 h | – | – |
| 3 | Pd(OAc)2 (5) | XPhos | Et3N | DMF | 10 h | – | – |
| 4 | Pd(OAc)2 (5) | XantPhos | Et3N | DMF | 10 h | – | – |
| 5 | Pd(OAc)2 (5) | DPEPhos | Et3N | DMF | 10 h | – | – |
| 6 | Pd2dba3 (5) | t-BuXPhos | DBU | DMF | 24 h | traces | – |
| 7 | Pd(OAc)2 (10) | t-BuXPhos | DBU | DMF | 24 h | 15 | 5 |
| 8c | Pd(OAc)2 (10) | t-BuXPhos | DBU | DMF | 10 h | 10 | – |
| 9c | Pd(OAc)2 (10) | t-BuXPhos | t-BuOK | DMF | 24 h | 22 | – |
| 10c | Pd(OAc)2 (10) | t-BuXPhos | Cs2CO3 | DMF | 24 h | traces | – |
| 11c | Pd(OAc)2 (10) | t-BuXPhos | DBU | dioxane | 24 h | 40 | 48 |
| 12c | Pd(OAc)2 (10) | XPhos | Cs2CO3 | dioxane | 10 h | 45 | 39 |
| 13d | Pd(OAc)2 (10) | DPEPhos | Cs2CO3 | dioxane | 10 h | 35 | 30 |
| 14d | Pd(OAc)2 (10) | SPhos | Cs2CO3 | toluene | 10 h | 20 | 55 |
aReaction conditions: o-phenylenediamine (1a, 0.46 mmol), dibromobenzene (2, 0.46 mmol), Pd catalytic system, base (2.5 equiv), DMF (5 mL), 110 °C, 24 h; o-(2-bromophenyl)aminoaniline (3a) and 5,10-dihydrophenazine (6) products were detected by TLC and 1H NMR, and yields determined after product isolation. bIsolated yields. c1.5 Equivalents of base were used. d1.2 Equivalents of base were used.
Interestingly, we noticed that in the initial one-pot reactions indicated in Table 1, the yields of the diarylamine 3a were better when Mo(CO)6 was present (compare entry 4, Table 1 to entry 7, Table 2). This might be due to (a) the fact that the temperature used for the reactions described in Table 1, involving Mo(CO)6 were 20 °C higher than the reactions described in Table 2 and/or (b) Mo(CO)6 acts as a co-catalyst. An investigative study was thus undertaken to elucidate the effect of the molybdenum reagent on the reaction using a simple model system consisting of aniline and bromobenzene (see Figure S1 and Table S1 in Supporting Information File 1). The experimental design was limited, in that we only monitored the reaction over a 90 min period. Contrary to what was originally believed the reaction without the Mo reagent gave better results during the first 90 min, but without additional data it is impossible to draw firm conclusions about this reaction, and thus further studies will be carried out in the near future.
After uncovering the optimum conditions to access o-(2-bromophenyl)aminoaniline via the B–H coupling reaction, it was decided to explore the carbonylative intramolecular cyclization of the intermediate 3a using different catalytic systems. To elucidate the role of the palladium catalyst in this process, we carried out the initial attempt under metal free-conditions using molybdenum hexacarbonyl (Mo(CO)6) as CO surrogate, in the presence of Et3N in DMF. The reaction was performed at 130 °C, as we believed that high temperature will promote the cyclization of the sterically hindered intermediate 3a, but no DBDAP was achieved under these conditions (entry 1, Table 3). Next, Pd(OAc)2 was employed under ligand-free conditions, but again the desired DBDAP product 4a could not be attained (entry 2, Table 3). Then, we performed the reaction in the presence of t-BuXPhos as ligand and in this case, only traces of the DBDAP 4a were obtained (entry 3, Table 3). When the reaction was carried out in the presence of DPEPhos (entry 4, Table 3), we were delighted to obtain the final dibenzodiazepine in 80% yield. Next, we tested another bidentate ligand, XantPhos, which led to the obtention of the desired product 4a in an excellent yield of 90% (entry 5, Table 3). This result implied that diphosphine ligands were essential for the success of this reaction. The reactivity of Co2CO8 as CO surrogate was also explored, in this case the reaction afforded the DBDAP product 4a in 55% yield (entry 6, Table 3). Molybdenum hexacarbonyl (Mo(CO)6), was shown to be a powerful CO surrogate in this carbonylative intramolecular cyclization. The efficacy of Mo(CO)6 is due to the energetically favorable dissociation of Mo(CO)n into Mo(CO)n−1 which was proven to be a highly exothermic reaction in the presence of metal catalysts especially after the dissociation of the first CO group [23].
Table 3: The intramolecular catalytic carbonylative cyclization conditions for o-(2-bromophenyl)aminoaniline.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-19-i8.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalytic system | CO surrogate | Yield 4ab (over 2 steps) |
| 1 | None | Mo(CO)6 | – |
| 2 | Pd(OAc)2 | Mo(CO)6 | – |
| 3 | Pd(OAc)2/t-BuXPhos | Mo(CO)6 | traces |
| 4 | Pd(OAc)2/DPEPhos | Mo(CO)6 | 80% (44%) |
| 5 | Pd(OAc)2/XantPhos | Mo(CO)6 | 90% (50%) |
| 6 | Pd(OAc)2/XantPhos | Co2(CO)8 | 55% (30%) |
aReaction conditions: o-(2-bromophenyl)aminoaniline (3a, 0.46 mmol), Pd(OAc)2 (10 mol %), ligand (30 mol %, Mo(CO)6 (1 equiv), Et3N (1 equiv) DMF (5 mL), 130 °C, 20 h. The reaction was monitored by TLC. bIsolated yields.
It should be noted that the best overall yield for the synthesis of 4a using the step-wise approach was 50% (Table 3).
Synthesis of dibenzodiazepinones via Chan–Lam amination/carbonylative coupling
Synthesis of o-(2-bromophenyl)aminoaniline via Chan–Lam C–N coupling
Inspired by the previously independently reported work by Chan [24] and Lam [25] and co-workers, we considered performing the reaction of o-phenylenediamine (1a) with 2-bromophenylboronic acid (7) in the presence of Cu(OAc)2, Et3N as base in DCM at 50 °C (entry 1, Table 4), and gratifingly under these conditions, the reaction afforded compound 3a in 48% yield. The Chan–Lam couplings were undertaken under an aerobic atmosphere which is an environmentally benign oxidant. Further screening using dioxane as solvent resulted in an increase in the yield to 55%, whilst DMF gave access to 3a in a lower yield (30%) (entries 2 and 3, Table 4). Next, we considered testing the performance of copper iodide (CuI, 20 mol %) as catalyst, in the presence of Et3N both in dioxane and DMF. These conditions resulted in the obtainment of the desired compound 3a in 59% and 35% yield, respectively (entries 4 and 5, Table 4). Increasing the reaction temperature to 100 °C resulted in a reduction of the reaction yield due to purported catalyst degradation (entry 6, Table 4). Then, we considered decreasing the amount of CuI to 10 mol % which led to a decrease of the reaction yield to 31% (entry 7, Table 4). Other bases such as dimethylaminopyridine (DMAP) and diisopropylethylamine (DIPEA) were tested under the previous reaction conditions but failed to improve the yield. In the presence of DMAP, no sign of the target compound was detected, while with DIPEA, 3a was obtained in 19% yield (entries 8 and 9, Table 4). Then, we tested CuSO4·5H2O in the presence of Et3N as the base in two different solvents DCM and dioxane, and both gave the desired compound in good yields of 60% and 50%, respectively (entries 10 and 11, Table 4).
Table 4: Optimization of the Chan–Lam coupling conditions between o-phenylenediamine (1a) and 2-bromophenylboronic acid (7).a
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-19-i9.svg?max-width=637&scale=1.0)
|
||||
| Entry | Copper source | Base | Solvent | Yield 3ab |
| 1 | Cu(OAc)2 | Et3N | DCM | 48% |
| 2 | Cu(OAc)2 | Et3N | dioxane | 55% |
| 3 | Cu(OAc)2 | Et3N | DMF | 30% |
| 4 | CuI | Et3N | dioxane | 59% |
| 5 | CuI | Et3N | DMF | 35% |
| 6c | CuI | Et3N | DMF | 23% |
| 7d | CuI | Et3N | DMF | 31% |
| 8 | CuI | DMAP | DMF | ND |
| 9 | CuI | DIPEA | DMF | 19% |
| 10 | CuSO4·5H2O | Et3N | dioxane | 60% |
| 11 | CuSO4·5H2O | Et3N | DCM | 50% |
aReaction conditions: o-phenylenediamine (1a, 0.46 mmol), 2-bromophenylboronic acid (7, 0.46 mmol, 1 equiv), copper catalyst (20 mol %), base (1.5 equiv), solvent (5 mL), 50 °C, 1–2 h. The reaction was monitored by TLC. bIsolated yield; ND: not detected cReaction performed at 100 °C. d10 mol % CuI were used.
Accessing the scope of the one-pot Chan–Lam/Pd-catalyzed carbonylative cyclization
Once the abovementioned conditions were obtained, we undertook a screening of the Chan–Lam/carbonylative synthesis of the DBDA in a one-pot manner. In the first reaction, o-phenylenediamine (1a) and 2-bromophenylboronic acid (7) were reacted in a pressure flask under an inert atmosphere using copper iodide (CuI) as the copper catalyst, Et3N, and Mo(CO)6 as CO surrogate in the presence of Pd(OAc)2/XantPhos as catalytic system for the carbonylative intramolecular cyclization. These conditions led to the obtainment of the compound 4a in 45% yield (entry 1, Table 5). Using dioxane as solvent, in the presence of the same catalytic system, afforded the desired structure 4a in moderate yield (37%, entry 2, Table 5). Changing the ligand to DPEPhos resulted in a slight increase of the yield of 4a, which was obtained in 40% (entry 3, Table 5). When the catalytic system PdCl2(NCCH3)/DPEPhos was employed, the product 4a was accessed in a lower yield of 18% (entry 4, Table 5). Under these reaction conditions, copper bromide was shown to be a good catalyst for this transformation, as it allowed the production of 4a in 40% yield (entry 5, Table 5). Then, we considered testing the performance of a different copper source Cu(OAc)2 under these reaction conditions using DMF and dioxane as solvents. In these cases, the final compound 4a was obtained in 38% and 41% yield, respectively (entries 6 and 7, Table 5). Next, we evaluated the performance of CuSO4·5H2O in dioxane and DMF, but lower yields were obtained (entries 8 and 9, Table 5). With the optimized conditions in hand, we tested the one pot Chan–Lam intramolecular cyclization with several other o-phenyldiamine derivatives, however, several impurities were obtained. In the hope of obtaining better yields (best obtained with the one pot method = 41%) we looked at the step-wise synthesis.
Table 5: Substrate scope of the one-pot synthesis of dibenzodiazepine using via Chan–Lam coupling/carbonylative intramolecular cyclization.a
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-19-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Copper source | Base | Solvent | Pd/ligand | Yield 4ab |
| 1 | CuI | Et3N | DMF | Pd(OAc)2/XantPhos | 45% |
| 2 | CuI | Et3N | dioxane | Pd(OAc)2/XantPhos | 37% |
| 3 | CuI | Et3N | DMF | Pd(OAc)2/DPEPhos | 40% |
| 4 | CuI | Et3N | DMF | PdCl2(NCCH3)2/XantPhos | 18% |
| 5 | CuBr | Et3N | DMF | Pd(OAc)2/Xantphos | 40% |
| 6 | Cu(OAc)2 | Et3N | DMF | Pd(OAc)2/DPEPhos | 38% |
| 7 | Cu(OAc)2 | Et3N | dioxane | Pd(OAc)2/DPEPhos | 41% |
| 8 | CuSO4.5H2O | Et3N | dioxane | Pd(OAc)2/DPEPhos | 18% |
| 9 | CuSO4.5H2O | Et3N | DMF | Pd(OAc)2/DPEPhos | traces |
aReaction conditions: o-phenylenediamine (1a, 0.5 mmol), 7 (0.5 mmol), Cu catalyst (20 mol %), base (0.6 mmol), solvent (5 mL), 50 °C, 1 h; Pd catalytic system (5 mol %), Mo(CO)6 (0.5 mmol) and base (0.5 mmol), 130 °C. bIsolated yields.
Synthesis of 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-ones via a stepwise Chan–Lam/carbonylative cyclization
After disclosing the optimal conditions for the Chan–Lam coupling, we screened different varieties of o-phenylenediamine derivatives. Overall, the o-phenylenene substrates bearing electron-donating substituents on the benzene ring proceeded smoothly under these conditions and led to the desired structures in moderate to good yields (Scheme 3). The reactions were generally regioselective, except in the case of 4-methyl-o-phenylenediamine (1b) and o-bromophenylboronic acid (7) which gave a mixture of products 3b and 3c in 60% yield (the ratio could not be determined). These were eventually separated and used in the cyclization step discussed below. The dimethylated o-phenylenediamine 1d, gave the desired compound 3d in 51% yield. A slight decrease in yield was observed in the presence of an ester (COOEt) substituent, which furnished compound 3g in 43% yield. Lower yields were observed in the case of electron-withdrawing substituents such as Cl and CF3 groups, which afforded the compounds 3e and 3f in 37% and 28% yields, respectively. The N-methylated precursor 1h was also tolerated by this system and afforded the desired compound 3h in 35% yield. Unfortunately, the reaction with 2-bromo-6-fluorophenylboronic acid (8) afforded the corresponding product 3i only in trace amounts.
![[1860-5397-20-19-i3]](/bjoc/content/inline/1860-5397-20-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the Chan–Lam coupling between o-phenylenediamines and 2-bromophenylboronic acids (please note products 3e and 3g contained some unidentified impurities that were impossible to remove via chromatography).
Scheme 3: Scope of the Chan–Lam coupling between o-phenylenediamines and 2-bromophenylboronic acids (please n...
With the o-(2-bromophenyl)aminoaniline derivatives in hand, we conducted the carbonylative intramolecular cyclization according to the previously disclosed conditions, in order to access the desired DBDAP structures. The unsubstituted DBDAP structure 4a was obtained in 80% under these conditions (Scheme 4). The methyl-substituted o-(2-bromophenyl)aminoanilines 3 obtained via Chan–Lam coupling could be efficiently separated and were subjected to the intramolecular carbonylative cyclization to yield the DBDAPs 4b and 4c in excellent yields of 95% and 93%, respectively. The dimethyl-DBDAP 4d could also be efficiently obtained under these conditions in 75% yield. The chloro-substituted DBDAP 4e, which is the intermediate to the antipsychotic drug clozapine, could also be obtained in good yield (this represented a formal synthesis to clozapine [26], if the procedure of Rao [27] is used, which entails heating 4e with 1-methylpiperidine and Ti(IV)Cl4, Scheme 4). Also compound 4a can be transformed to Hügel's 1,2,3-triazole-DBDAP using the methodology described in their report (Scheme 4) [5]. The CF3- and COOEt-substituted DBDAs 4f and 4g were obtained with a slightly decreased yield of 55% and 40%, respectively. The N-methyldibenzodiazepine 4h could be accessed, but only in trace quantity. It should be noted that the stepwise approach was slightly better than the one-pot approach in the case of the synthesis 4a (47% vs 41%).
![[1860-5397-20-19-i4]](/bjoc/content/inline/1860-5397-20-19-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Scope of the synthesis of DBDAPs. Please note that product 4g contained some unidentified impurities that were impossible to remove via chromatography.
Scheme 4: Scope of the synthesis of DBDAPs. Please note that product 4g contained some unidentified impuritie...
Our mechanistic proposal is based on the information in previous reports by the groups of Bose [28], Watson [29], and Stahl [30]. Mechanistically, under basic conditions, the reaction is triggered by copper-catalyzed activation of o-phenylenediamine (1a), followed by the oxygen-promoted insertion of the phenylboronic acid coupling partner 7 to deliver intermediate II that undergoes reductive elimination to give diarylamine 3a along with regeneration of the copper catalyst (Scheme 5). Then, a palladium-promoted oxidative addition of the C–Br bond takes places to deliver palladium species III. Then the insertion of CO that is released by Mo(CO)6, should afford intermediate IV that undergoes a base-promoted intramolecular cyclization via nucleophilic attack of the amine [31]. Finally, the dibenzodiazepinone 4a would be obtained through reductive elimination of the palladium catalyst.
![[1860-5397-20-19-i5]](/bjoc/content/inline/1860-5397-20-19-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have reported two one-pot pathways and two step-wise pathways to access dibenzodiazepinone (DBDAP) derivatives via copper-catalyzed Chan–Lam amination/carbonylative cyclization and Buchwald–Hartwig amination/carbonylative cyclization and their step-wise counterparts. Although the one-pot method worked for one example in both cases (but in one case it gave the DBDAP enol form), it failed to work for other substrates, and for that reason we had to rely on the step-wise approach. The more efficient method to access the diamine intermediates 3 was via the Chan–Lam amination (milder conditions, cheaper, earth-abundant catalyst, no expensive ligand requirement) as the Buchwald–Hartwig amination required harsher conditions and an expensive metal catalyst, and also gave an unwanted phenazine side product. The sequential stepwise Chan–Lam amination/carbonylative cyclization afforded a number of DBDAP products, showing good functional group tolerance and giving the final products in good yields. In terms of the overall best efficiency, it also would appear that the step-wise Chan–Lam/Pd-catalyzed carbonylative cyclization was slightly better than the one-pot method. The most important of which was the chloro-containing DBDAP 4e that can be used to synthesize the antipsychotic drug clozapine (see above), a triazole-hybrid with anticancer properties, and can easily be used as the key part in the synthesis of other drugs like dibenzepine and biologically active natural products such as BU-4664L. We are currently looking at this methodology to access some of these targets, including the agrochemical boscalid.
Experimental
Synthesis of o-(2-bromophenyl)aminoaniline (3a)
Via Buchwald–Hartwig coupling: o-Phenylenediamine (1a, 0.05g, 1 equiv, 0.46 mmol) was added to a Radleys reaction tube (a Radleys® 12 position carousel reactor station was used) under N2 and dissolved in dry dioxane (5 mL). Next, (0.055 mL, 0.46 mmol) of 1,2-dibromobenzene (2) was added to the reaction mixture, followed by the addition of Pd(OAc)2 (0.01 g, 0.046 mmol), XPhos (0.032 g, 0.069 mmol), and Cs2CO3 (0.18 g, 0. 56 mmol, 1,2 equiv). The resulting reaction mixture was allowed to stir at 100 °C. The reaction was left stirring for several hours, followed by TLC. After consumption of the starting material (verified through TLC). The reaction was allowed to cool and filtered through a celite pad to remove the residual catalyst and base. The solvent was then evaportated under reduced pressure and the crude was purified by flash chromatography (hexane/AcOEt 9:1), to yield o-(2-bromophenyl)aminoaniline (3) compound as a purple oil (0.057 g, 47% yield).
Via Chan–Lam coupling: o-Phenylenediamine (1a, 0.05 g, 1 equiv, 0.46 mmol) was added to a round-bottomed flask and dissolved in dry dioxane (5 mL). Next, 2-bromophenyl)boronic acid (7, 0.092 g, 1 equiv,0.46 mmol) was added, followed by the addition of Et3N (0.07 mL, 0.055 mmol), CuI (0.018 g, 0.092 mmol, 20 mol %), and molecular sieves 3 Å. The reaction was left stirring at room temperature for several hours, and monitored by TLC. After consumption of the starting material (verified through TLC), the reaction mixture was filtered through a celite pad to remove the residual catalyst and molecular sieves. The volatiles were then evaporated under reduced pressure and the crude was purified by flash chromatography (hexane/AcOEt 9:1), to yield o-(2-bromophenyl)aminoaniline (3a) as a purple oil (0.07 g, 59% yield).1H NMR (CDCl3, 400 MHz) δ 4.00 (s, NH2, 2H), 5.76 (s, NH, 1H), 6.59–6.61 (d, J = 8 Hz, Ar, 1H), 6.65–6.69 (t, J = 8 Hz, Ar, 1H), 6.79–6.83 (t, J = 8 Hz, Ar, 1H), 6.85–6.87 (d, J = 8 Hz, Ar, 1H), 7.09–7.13 (m, Ar, 3H), 7.49–7.51 (d, J = 8 Hz, Ar, 1H); 13C NMR (CDCl3, 100 MHz) δ 110.42, 114.41, 116.47, 119.48, 119.70, 127.00, 127.04, 128.39, 132.62, 142.45, 143.03; HRESIMS (m/z): [M + H+] calcd for C12H11BrN2, 263,0184; found, 263.0178.
Synthesis of 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-one (4a)
o-(2-Bromophenyl)aminoaniline (3a, 0.05 g, 0.19 mmol) was added to a Radley’s® 12 position carousel reactor tube to which DMF, then Pd(OAc)2 (4.26 mg, 0.019 mmol), DPEPhos (30 mg, 0.057 mmol), Mo(CO)6 (50 mg, 1 equiv, 0.19 mmol), and Et3N (0.026 mL, 0.19 mmol) were added. The reaction mixture was then stirred at 130 °C under a nitrogen atmosphere. After completion of the reaction, as determined by TLC, the reaction mixture was allowed to cool to room temperature. The mixture was filtered through a pad of celite and washed with DCM, then, the solvent was evaporated under reduced pressure to give a crude mixture. Further purification by flash chromatography (hexane/AcOEt 1:1), gave the desired compound 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-one (4a) as a yellow solid yield (0.032 g, 80%). Mp 249–251 °C; 1H NMR (DMSO-d6, 400 MHz) δ 6.87–7.00 (m, Ar, 6H), 7.31–7.35 (t, J = 8 Hz, Ar, 1H), 7.66–7.68 (d, J = 8 Hz, Ar,1H), 7.84 (s, Ar, 1H), 9.85 (s, Ar, 1H); 13C NMR (CDCl3, 100 MHz) δ 119.52, 120.23, 121.17, 121.73, 123.24, 123.40, 124.95, 130.29, 132.56, 133.67, 140.43, 150.92, 168.40; ESIMS (m/z): 221.12 [M + H+].
Supporting Information
| Supporting Information File 1: Experimental procedures and spectral data (NMR, mass spectra) and key kinetic studies. | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Khokhar, J. Y.; Henricks, A. M.; Sullivan, E. D. K.; Green, A. I. Adv. Pharmacol. (San Diego, CA, U. S.) 2018, 82, 137–162. doi:10.1016/bs.apha.2017.09.009
Return to citation in text: [1] -
Jafari, S.; Fernandez‐Enright, F.; Huang, X.-F. J. Neurochem. 2012, 120, 371–384. doi:10.1111/j.1471-4159.2011.07590.x
Return to citation in text: [1] -
Cao, K.; Yan, J.; Yan, F.; Yin, T. Mol. Diversity 2021, 25, 1111–1122. doi:10.1007/s11030-020-10051-z
Return to citation in text: [1] -
Miyanaga, S.; Sakurai, H.; Saiki, I.; Onaka, H.; Igarashi, Y. Bioorg. Med. Chem. Lett. 2010, 20, 963–965. doi:10.1016/j.bmcl.2009.12.055
Return to citation in text: [1] -
Praveen Kumar, C.; Reddy, T. S.; Mainkar, P. S.; Bansal, V.; Shukla, R.; Chandrasekhar, S.; Hügel, H. M. Eur. J. Med. Chem. 2016, 108, 674–686. doi:10.1016/j.ejmech.2015.12.007
Return to citation in text: [1] [2] -
Minucci, S.; Pelicci, P. G. Nat. Rev. Cancer 2006, 6, 38–51. doi:10.1038/nrc1779
Return to citation in text: [1] -
De Clercq, D. J. H.; Heppner, D. E.; To, C.; Jang, J.; Park, E.; Yun, C.-H.; Mushajiang, M.; Shin, B. H.; Gero, T. W.; Scott, D. A.; Jänne, P. A.; Eck, M. J.; Gray, N. S. ACS Med. Chem. Lett. 2019, 10, 1549–1553. doi:10.1021/acsmedchemlett.9b00381
Return to citation in text: [1] -
Aniban, X.; Mamidala, S.; Burke, A. J. Eur. J. Org. Chem. 2018, 6743–6753. doi:10.1002/ejoc.201801304
Return to citation in text: [1] -
Guo, W.; Zhao, M.; Tan, W.; Zheng, L.; Tao, K.; Fan, X. Org. Chem. Front. 2019, 6, 2120–2141. doi:10.1039/c9qo00283a
Return to citation in text: [1] -
Chen, J.-Q.; Li, J.-H.; Dong, Z.-B. Adv. Synth. Catal. 2020, 362, 3311–3331. doi:10.1002/adsc.202000495
Return to citation in text: [1] -
Schoenberg, A.; Heck, R. F. J. Org. Chem. 1974, 39, 3327–3331. doi:10.1021/jo00937a004
Return to citation in text: [1] -
Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 4114–4133. doi:10.1002/anie.200900013
Return to citation in text: [1] -
Tsvelikhovsky, D.; Buchwald, S. L. J. Am. Chem. Soc. 2011, 133, 14228–14231. doi:10.1021/ja206229y
Return to citation in text: [1] -
Zhang, Q.-Y.; Wang, X.-J.; Tian, Y.-L.; Qi, J.-G.; Li, C.; Yin, D.-L. Chin. Chem. Lett. 2013, 24, 825–828. doi:10.1016/j.cclet.2013.04.049
Return to citation in text: [1] -
Laha, J. K.; Manral, N.; Hunjan, M. K. New J. Chem. 2019, 43, 7339–7343. doi:10.1039/c9nj00539k
Return to citation in text: [1] -
Åkerbladh, L.; Odell, L. R.; Larhed, M. Synlett 2019, 30, 141–155. doi:10.1055/s-0037-1610294
Return to citation in text: [1] -
Odell, L. R.; Russo, F.; Larhed, M. Synlett 2012, 23, 685–698. doi:10.1055/s-0031-1290350
Return to citation in text: [1] -
Peixoto, D.; Locati, A.; Marques, C. S.; Goth, A.; Ramalho, J. P. P.; Burke, A. J. RSC Adv. 2015, 5, 99990–99999. doi:10.1039/c5ra19599c
Return to citation in text: [1] -
Wan, Y.; Alterman, M.; Larhed, M.; Hallberg, A. J. Org. Chem. 2002, 67, 6232–6235. doi:10.1021/jo025965a
Return to citation in text: [1] -
Oseghale, C. O.; Onisuru, O. R.; Fapojuwo, D. P.; Mogudi, B. M.; Molokoane, P. P.; Maqunga, N. P.; Meijboom, R. RSC Adv. 2021, 11, 26937–26948. doi:10.1039/d1ra05177f
Return to citation in text: [1] [2] -
Wannberg, J.; Larhed, M. J. Org. Chem. 2003, 68, 5750–5753. doi:10.1021/jo034382d
Return to citation in text: [1] -
Hussain, N.; Chhalodia, A. K.; Ahmed, A.; Mukherjee, D. ChemistrySelect 2020, 5, 11272–11290. doi:10.1002/slct.202003395
Return to citation in text: [1] -
Dupont, C.; Wan, X.; Petukhov, M.; Krüger, P. Int. J. Quantum Chem. 2014, 114, 1630–1635. doi:10.1002/qua.24744
Return to citation in text: [1] -
Chan, D. M. T. Tetrahedron Lett. 1996, 37, 9013–9016. doi:10.1016/s0040-4039(96)02116-8
Return to citation in text: [1] -
Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/s0040-4039(98)00504-8
Return to citation in text: [1] -
Nucifora, F. C., Jr.; Mihaljevic, M.; Lee, B. J.; Sawa, A. Neurotherapeutics 2017, 14, 750–761. doi:10.1007/s13311-017-0552-9
Return to citation in text: [1] -
Venkat Rao, S. Arabian J. Chem. 2020, 13, 6040–6043. doi:10.1016/j.arabjc.2020.05.003
Return to citation in text: [1] -
Bose, S.; Dutta, S.; Koley, D. ACS Catal. 2022, 12, 1461–1474. doi:10.1021/acscatal.1c04479
Return to citation in text: [1] -
Vantourout, J. C.; Miras, H. N.; Isidro-Llobet, A.; Sproules, S.; Watson, A. J. B. J. Am. Chem. Soc. 2017, 139, 4769–4779. doi:10.1021/jacs.6b12800
Return to citation in text: [1] -
King, A. E.; Ryland, B. L.; Brunold, T. C.; Stahl, S. S. Organometallics 2012, 31, 7948–7957. doi:10.1021/om300586p
Return to citation in text: [1] -
Shen, C.; Neumann, H.; Wu, X.-F. Green Chem. 2015, 17, 2994–2999. doi:10.1039/c5gc00427f
Return to citation in text: [1]
| 30. | King, A. E.; Ryland, B. L.; Brunold, T. C.; Stahl, S. S. Organometallics 2012, 31, 7948–7957. doi:10.1021/om300586p |
| 31. | Shen, C.; Neumann, H.; Wu, X.-F. Green Chem. 2015, 17, 2994–2999. doi:10.1039/c5gc00427f |
| 1. | Khokhar, J. Y.; Henricks, A. M.; Sullivan, E. D. K.; Green, A. I. Adv. Pharmacol. (San Diego, CA, U. S.) 2018, 82, 137–162. doi:10.1016/bs.apha.2017.09.009 |
| 2. | Jafari, S.; Fernandez‐Enright, F.; Huang, X.-F. J. Neurochem. 2012, 120, 371–384. doi:10.1111/j.1471-4159.2011.07590.x |
| 6. | Minucci, S.; Pelicci, P. G. Nat. Rev. Cancer 2006, 6, 38–51. doi:10.1038/nrc1779 |
| 16. | Åkerbladh, L.; Odell, L. R.; Larhed, M. Synlett 2019, 30, 141–155. doi:10.1055/s-0037-1610294 |
| 17. | Odell, L. R.; Russo, F.; Larhed, M. Synlett 2012, 23, 685–698. doi:10.1055/s-0031-1290350 |
| 5. | Praveen Kumar, C.; Reddy, T. S.; Mainkar, P. S.; Bansal, V.; Shukla, R.; Chandrasekhar, S.; Hügel, H. M. Eur. J. Med. Chem. 2016, 108, 674–686. doi:10.1016/j.ejmech.2015.12.007 |
| 18. | Peixoto, D.; Locati, A.; Marques, C. S.; Goth, A.; Ramalho, J. P. P.; Burke, A. J. RSC Adv. 2015, 5, 99990–99999. doi:10.1039/c5ra19599c |
| 4. | Miyanaga, S.; Sakurai, H.; Saiki, I.; Onaka, H.; Igarashi, Y. Bioorg. Med. Chem. Lett. 2010, 20, 963–965. doi:10.1016/j.bmcl.2009.12.055 |
| 14. | Zhang, Q.-Y.; Wang, X.-J.; Tian, Y.-L.; Qi, J.-G.; Li, C.; Yin, D.-L. Chin. Chem. Lett. 2013, 24, 825–828. doi:10.1016/j.cclet.2013.04.049 |
| 3. | Cao, K.; Yan, J.; Yan, F.; Yin, T. Mol. Diversity 2021, 25, 1111–1122. doi:10.1007/s11030-020-10051-z |
| 15. | Laha, J. K.; Manral, N.; Hunjan, M. K. New J. Chem. 2019, 43, 7339–7343. doi:10.1039/c9nj00539k |
| 10. | Chen, J.-Q.; Li, J.-H.; Dong, Z.-B. Adv. Synth. Catal. 2020, 362, 3311–3331. doi:10.1002/adsc.202000495 |
| 12. | Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 4114–4133. doi:10.1002/anie.200900013 |
| 9. | Guo, W.; Zhao, M.; Tan, W.; Zheng, L.; Tao, K.; Fan, X. Org. Chem. Front. 2019, 6, 2120–2141. doi:10.1039/c9qo00283a |
| 13. | Tsvelikhovsky, D.; Buchwald, S. L. J. Am. Chem. Soc. 2011, 133, 14228–14231. doi:10.1021/ja206229y |
| 8. | Aniban, X.; Mamidala, S.; Burke, A. J. Eur. J. Org. Chem. 2018, 6743–6753. doi:10.1002/ejoc.201801304 |
| 7. | De Clercq, D. J. H.; Heppner, D. E.; To, C.; Jang, J.; Park, E.; Yun, C.-H.; Mushajiang, M.; Shin, B. H.; Gero, T. W.; Scott, D. A.; Jänne, P. A.; Eck, M. J.; Gray, N. S. ACS Med. Chem. Lett. 2019, 10, 1549–1553. doi:10.1021/acsmedchemlett.9b00381 |
| 11. | Schoenberg, A.; Heck, R. F. J. Org. Chem. 1974, 39, 3327–3331. doi:10.1021/jo00937a004 |
| 20. | Oseghale, C. O.; Onisuru, O. R.; Fapojuwo, D. P.; Mogudi, B. M.; Molokoane, P. P.; Maqunga, N. P.; Meijboom, R. RSC Adv. 2021, 11, 26937–26948. doi:10.1039/d1ra05177f |
| 22. | Hussain, N.; Chhalodia, A. K.; Ahmed, A.; Mukherjee, D. ChemistrySelect 2020, 5, 11272–11290. doi:10.1002/slct.202003395 |
| 19. | Wan, Y.; Alterman, M.; Larhed, M.; Hallberg, A. J. Org. Chem. 2002, 67, 6232–6235. doi:10.1021/jo025965a |
| 20. | Oseghale, C. O.; Onisuru, O. R.; Fapojuwo, D. P.; Mogudi, B. M.; Molokoane, P. P.; Maqunga, N. P.; Meijboom, R. RSC Adv. 2021, 11, 26937–26948. doi:10.1039/d1ra05177f |
| 21. | Wannberg, J.; Larhed, M. J. Org. Chem. 2003, 68, 5750–5753. doi:10.1021/jo034382d |
| 28. | Bose, S.; Dutta, S.; Koley, D. ACS Catal. 2022, 12, 1461–1474. doi:10.1021/acscatal.1c04479 |
| 29. | Vantourout, J. C.; Miras, H. N.; Isidro-Llobet, A.; Sproules, S.; Watson, A. J. B. J. Am. Chem. Soc. 2017, 139, 4769–4779. doi:10.1021/jacs.6b12800 |
| 27. | Venkat Rao, S. Arabian J. Chem. 2020, 13, 6040–6043. doi:10.1016/j.arabjc.2020.05.003 |
| 5. | Praveen Kumar, C.; Reddy, T. S.; Mainkar, P. S.; Bansal, V.; Shukla, R.; Chandrasekhar, S.; Hügel, H. M. Eur. J. Med. Chem. 2016, 108, 674–686. doi:10.1016/j.ejmech.2015.12.007 |
| 25. | Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/s0040-4039(98)00504-8 |
| 26. | Nucifora, F. C., Jr.; Mihaljevic, M.; Lee, B. J.; Sawa, A. Neurotherapeutics 2017, 14, 750–761. doi:10.1007/s13311-017-0552-9 |
| 23. | Dupont, C.; Wan, X.; Petukhov, M.; Krüger, P. Int. J. Quantum Chem. 2014, 114, 1630–1635. doi:10.1002/qua.24744 |
| 24. | Chan, D. M. T. Tetrahedron Lett. 1996, 37, 9013–9016. doi:10.1016/s0040-4039(96)02116-8 |
© 2024 Moutayakine and Burke; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.