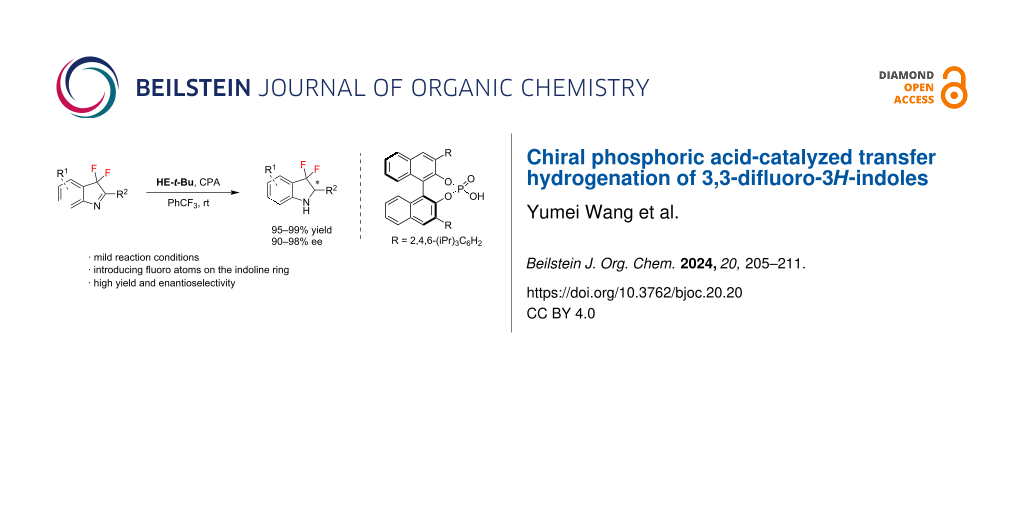
Abstract
A convenient and efficient method for the synthesis of optically active difluoro-substituted indoline derivatives starting from the corresponding 3H-indoles by chiral phosphoric acid-catalyzed transfer hydrogenation was developed. Using Hantzsch ester as the hydrogen source under mild reaction conditions, the target products can be obtained with excellent yield and enantioselectivity.

Graphical Abstract
Introduction
The introduction of fluoro atoms into organic molecules can alter their lipophilicity, solubility, metabolic stability, and increase drug activity by affecting drug receptor interactions [1]. Therefore, replacing hydrogen with one or more fluoro atoms has beneficial effects on therapeutic efficacy and pharmacological activity [2]. For example, flindokalner is a potassium channel opener (Figure 1) [3]. JAB-3068 is a promising SHP2 inhibitor that has entered phase II clinical trials for the treatment of solid tumors and has been approved by the FDA as a rare drug for treating esophageal cancer (Figure 1) [4]. Among the fluoroalkyl moieties, the geminal difluoromethylene group has showcased its beneficial properties as an isostere of polar functional groups [5,6].
![[1860-5397-20-20-1]](/bjoc/content/figures/1860-5397-20-20-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of bioactive fluorinated indole derivatives.
Figure 1: Structures of bioactive fluorinated indole derivatives.
Chiral indoline is an important member of the class of nitrogen-containing heterocyclic compounds that often exhibits various pharmaceutical activities and exists in many natural products [7,8]. The enantioselective synthesis of chiral indolines has received great attention in organic synthesis. Various methods [9], including reductive hydrogenation [10,11], kinetic resolution [12-14], functionalization of indole [15], and de novo construction of chiral 2-substituted indolines, have been developed [16-20]. In recent years, the metal-catalyzed asymmetric hydrogenation of indoles to synthesize chiral indolines has been widely studied (Scheme 1a) [21,22]. Representative examples include Ir- or Ru-catalyzed asymmetric hydrogenation of 2,3,3-trisubstituted 3H-indole [23,24]. Generally, these methods employ precious metals and/or relatively strict reaction conditions (up to 150 bar H2). In 2022, Liu’s group reported an asymmetric hydrogenation of 3H-indoles catalyzed by a chiral Mn complex, which showed good yield and enantioselectivity [25]. In addition to metal catalysis for the enantioselective reduction, asymmetric organocatalysis using chiral phosphoric acids has also been studied (Scheme 1b) [26-28]. In 2010, Magnus Rueping and his co-workers developped an enantioselective Brønsted acid-catalyzed transfer hydrogenation of 3H-indoles [29]. In 2020, Song and Yu successfully applied a new chiral Brønsted acid, synthesized in situ from a chiral boron phosphate complex with water, for asymmetric indole reduction (Scheme 1b) [30]. The mild reaction conditions, low catalyst loading, and high enantioselectivity rendered this transformation an attractive approach to synthesize optically active indolines. However, these asymmetric reduction studies focused on alkyl or aryl-substituted 3H-indoles whereas the synthesis of chiral difluorinated indole derivatives could have potential applications in pharmaceutical chemistry. Herein, an organocatalyzed transfer hydrogenation of 3,3-difluoro-3H-indoles to obtain fluorinated 3H-indolines was developed (Scheme 1c). With this method, a variety of chiral 3,3-difluoroindolines were synthesized in high yield and enantioselectivity under mild reaction conditions.
![[1860-5397-20-20-i1]](/bjoc/content/inline/1860-5397-20-20-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of chiral indolines via asymmetric reduction.
Scheme 1: Synthesis of chiral indolines via asymmetric reduction.
Results and Discussion
We conducted a preliminary exploration of the reaction using 3,3-difluoro-2-(phenylethynyl)-3H-indole (1a) as the model substrate, Hantzsch ester (HE-Et) as the hydrogen source, and BINOL-derived chiral phosphoric acids (CPA) as the catalyst (Table 1). With chiral phosphoric acid CPA-1, the transfer hydrogenation reaction proceeded well in PhCF3 at room temperature and the target product 2a was obtained in 98% yield with 20% ee after 12 h (Table 1, entry 1). Then, the effect of steric hindrance of the CPA catalyst and solvents on the stereochemistry of this transfer hydrogenation were investigated in detail. Among various 3,3’-disubstituted CPA catalysts (Table 1, entries 2–6), chiral phosphoric acid CPA-6 containing 2,4,6-triisopropylphenyl-substituents at the 3,3’-positions of the binaphthyl skeleton performed best giving the target product in 99% yield with 91% ee (Table 1, entry 6). This suggested, that the steric hindrance of the CPA catalyst at the 3,3’-position is important for achieving high selectivity. Also, an obvious solvent effect on the enantioselectivity was observed (Table 1, entries 7–10). Very low ee values of product 2a were detected when the reaction was performed in DMSO or MeOH (Table 1, entries 7 and 8), while using DCE or toluene as the solvent the enantioselectivity dropped significantly (Table 1, entries 9 and 10).
Table 1: Reaction optimization studies.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-20-i4.svg?max-width=637&scale=1.0)
|
||||
| entry | chiral phosphoric acid | solvent | yield (%) | ee (%) |
| 1 | CPA-1 | PhCF3 | 98 | 20 |
| 2 | CPA-2 | PhCF3 | 99 | 24 |
| 3 | CPA-3 | PhCF3 | 99 | 67 |
| 4 | CPA-4 | PhCF3 | 99 | 40 |
| 5 | CPA-5 | PhCF3 | 99 | 76 |
| 6 | CPA-6 | PhCF3 | 99 | 91 |
| 7 | CPA-6 | DMSO | 99 | 7 |
| 8 | CPA-6 | MeOH | 99 | 7 |
| 9 | CPA-6 | DCE | 95 | 82 |
| 10 | CPA-6 | toluene | 99 | 85 |
aReaction conditions: 1a (0.1 mmol, 1.0 equiv), Hantzsch diethyl ester (1.5 equiv), CPA (10 mol %), solvent (1 mL), rt, under N2 atmosphere, 12 h. The yield was determined by 19F NMR spectroscopy and the ee value was determined by chiral HPLC. DMSO: dimethyl suifoxide; DCE: 1,2-dichloroethane.
To further improve the enantioselectivity of this CPA-catalyzed transfer hydrogenation, we next explored the effect of the alcohol part of Hantzsch esters (Table 2). The experimental results showed that the ee value of product 2a increased as the steric hinderance of Hantzsch ester raised (Table 2, entries 1–3) and switching from ethyl to tert-butyl esters the desired product was obtained in excellent yield and enantioselectivity (Table 2, entry 3). Subsequently, we investigated the effect of the amounts of HE-t-Bu and chiral phosphoric acid on the reaction outcome. When the amount of HE-t-Bu was decreased, the reaction yield dropped (Table 2, entry 4). On the other hand, reducing the amount of chiral phosphoric acid to 1 mol % or the reaction time to 3 hours, still good reaction results were observed (Table 2, entries 5 and 6). However, the enantioselectivity decreased when the reaction temperature was reduced to 0 °C (Table 2, entry 7).
Table 2: The effect of Hantzsch esters and other reaction parameters.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-20-i5.svg?max-width=637&scale=1.0)
|
|||
| entry | Hantzsch ester | yield (%)b | ee (%) |
| 1 | HE-Me (1.5 equiv) | 99 | 83 |
| 2 | HE-Et (1.5 equiv) | 99 | 91 |
| 3 | HE-t-Bu (1.5 equiv) | 99 | 96 |
| 4 | HE-t-Bu (1.0 equiv) | 88 | 96 |
| 5b | HE-t-Bu (1.5 equiv) | 99 | 96 |
| 6b,c | HE-t-Bu (1.5 equiv) | 99 | 96 |
| 7c,d | HE-t-Bu (1.5 equiv) | 99 | 85 |
aReaction conditions: 1a (0.1 mmol, 1.0 equiv), Hantzsch ester (1.5 equiv), CPA-6 (1 mol %), PhCF3 (1 mL), rt, under N2 atmosphere,12 h. The yield was determined by 19F NMR spectroscopy and the ee values were determined by chiral HPLC. b1 mol % of CPA-6 was used. cThe reaction time was 3 h. dThe reaction temperature was 0 °C.
With the optimal reaction conditions in hand, the substrate range of 2-alkynyl-3,3-difluoro-3H-indoles 1 for this transfer hydrogenation reaction was investigated (Scheme 2). Fluoro-, chloro-, and bromo-substituted 3,3-difluoro-2-(phenylethynyl)-3H-indoles were well tolerated, providing the chiral indolines 2b–e in high yields and ee values. Various 2-alkynyl-3,3-difluoro-3H-indoles bearing electron-donating and electron-withdrawing groups at the meta- (2f and 2g), para- (2h and 2m) or ortho- (2n and 2o) position of the aryl ring smoothly underwent this asymmetric reduction, affording the desired indolines in 95–99% yield and 90–96% ee within 3 hours. Replacing the 3,3-difluoro substituents by two methyl groups in the starting indole as well as the alkyne part by a phenyl group, the reaction still gave good results (2p). However, when using 3,3-difluoro-2-(naphthalen-2-ylethynyl)-3H-indole or 3,3-difluoro-2-phenyl-3H-indole as the substrate, the generated indoles underwent fast HF elimination/aromatization and finally gave indole derivatives (2q and 2r) in almost quantitative yields.
![[1860-5397-20-20-i2]](/bjoc/content/inline/1860-5397-20-20-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of 3,3-difluoro-3H-indoles.
Scheme 2: Substrate scope of 3,3-difluoro-3H-indoles.
To examine the efficiency and practicability of this approach, a 2 mmol scale experiment of the asymmetric transfer hydrogenation of 1a was carried out (Scheme 3). Under the standard reaction conditions, 0.5 gram (98% yield) of chiral difluorinated indoline 2a was obtained with 95% ee.
![[1860-5397-20-20-i3]](/bjoc/content/inline/1860-5397-20-20-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on previous studies [31], a mechanism of the CPA-catalyzed transfer hydrogenation reaction was proposed (Figure 2). The activation of 3,3-difluoro-substituted 3H-indole 1 by protonation through the Brønsted acid generates the iminium A. Subsequent hydrogen transfer from the Hantzsch ester gives the chiral amine 2 and pyridinium salt B. The CPA catalyst is regenerated from salt B through proton transfer. We deduced that the steric repulsion between the bulky 2,4,6-triisopropylphenyl-substitutents in the chiral phosphoric acid CPA-6 and the carboxylic ester group of the Hantzsch ester hydrogen donor contribute to the high enantioselectivity of the reaction. The role of fluorine and alkyne in the reaction should be close to the gem-dimethyl moiety and the phenyl group in the previous research [32].
![[1860-5397-20-20-2]](/bjoc/content/figures/1860-5397-20-20-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Proposed mechanism for the transfer hydrogenation reaction.
Figure 2: Proposed mechanism for the transfer hydrogenation reaction.
Conclusion
In summary, we developed a convenient method for the synthesis of chiral difluoroindoline compounds for the first time. With a chiral phosphoric acid as a Brønsted acid catalyst and Hantzsch ester as the hydrogen source, a series of 3,3-difluoro-substituted 3H-indoles underwent asymmetric transfer hydrogenation under mild reaction conditions, giving the target products with excellent yields and optical purity.
Experimental
General procedure: a 4 mL sample bottle was charged with 3,3-difluoro-substituted 3H-indole 1 (0.1 mmol, 1.0 equiv), Hantzsch ester (HE-t-Bu, 42.0 mg, 0.15 mmol, 1.5 equiv), and chiral phosphoric acid (CPA-6, 0.75 mg, 0.001 mmol, 1 mol %). Then, PhCF3 (1 mL) was added in a glove box under N2 atmosphere and the reaction mixture was stirred at room temperature for 3 h. After concentrating the mixture, the residue was purified by column chromatography on silica gel using a mixture of petroleum ether/ethyl acetate 30:1 (v/v) as the eluent to afford products 2. The yields were determined by 19F NMR spectroscopy and the ee values were determined by chiral HPLC.
Supporting Information
| Supporting Information File 1: Full experimental details and characterization data of all compounds. | ||
| Format: PDF | Size: 7.7 MB | Download |
References
-
Gupta, S. P. Lett. Drug Des. Discovery 2019, 16, 1089–1109. doi:10.2174/1570180816666190130154726
Return to citation in text: [1] -
Zhang, L.; Zhang, X.; Cui, Y.; Yang, C. Synthesis 2021, 53, 3815–3826. doi:10.1055/a-1509-8624
Return to citation in text: [1] -
Young, B. L.; Cooks, R. G.; Madden, M. C.; Bair, M.; Jia, J.; Aubry, A.-F.; Miller, S. A. J. Pharm. Biomed. Anal. 2007, 43, 1602–1608. doi:10.1016/j.jpba.2006.12.027
Return to citation in text: [1] -
Ma, C.; Gao, P.-L.; Chu, J.; Wu, X.-P.; Wen, C.-W.; Kang, D.; Bai, J.-L.; Pei, X.-Y. Novel heterocyclic derivatives useful as shp2 inhibitors. International Patent Application WO2017/211303 A1, Dec 14, 2017.
Return to citation in text: [1] -
Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Chem. – Eur. J. 2015, 21, 12836–12865. doi:10.1002/chem.201501475
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2011, 54, 2529–2591. doi:10.1021/jm1013693
Return to citation in text: [1] -
Famiglini, V.; La Regina, G.; Coluccia, A.; Masci, D.; Brancale, A.; Badia, R.; Riveira-Muñoz, E.; Esté, J. A.; Crespan, E.; Brambilla, A.; Maga, G.; Catalano, M.; Limatola, C.; Formica, F. R.; Cirilli, R.; Novellino, E.; Silvestri, R. J. Med. Chem. 2017, 60, 6528–6547. doi:10.1021/acs.jmedchem.6b01906
Return to citation in text: [1] -
Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Eur. J. Med. Chem. 2019, 183, 111691. doi:10.1016/j.ejmech.2019.111691
Return to citation in text: [1] -
Hua, T.-B.; Xiao, C.; Yang, Q.-Q.; Chen, J.-R. Chin. Chem. Lett. 2020, 31, 311–323. doi:10.1016/j.cclet.2019.07.015
Return to citation in text: [1] -
Duan, Y.; Li, L.; Chen, M.-W.; Yu, C.-B.; Fan, H.-J.; Zhou, Y.-G. J. Am. Chem. Soc. 2014, 136, 7688–7700. doi:10.1021/ja502020b
Return to citation in text: [1] -
Núñez-Rico, J. L.; Fernández-Pérez, H.; Vidal-Ferran, A. Green Chem. 2014, 16, 1153–1157. doi:10.1039/c3gc42132e
Return to citation in text: [1] -
Saito, K.; Akiyama, T. Angew. Chem., Int. Ed. 2016, 55, 3148–3152. doi:10.1002/anie.201510692
Return to citation in text: [1] -
Murray, J. I.; Flodén, N. J.; Bauer, A.; Fessner, N. D.; Dunklemann, D. L.; Bob‐Egbe, O.; Rzepa, H. S.; Bürgi, T.; Richardson, J.; Spivey, A. C. Angew. Chem., Int. Ed. 2017, 56, 5760–5764. doi:10.1002/anie.201700977
Return to citation in text: [1] -
Krasnov, V. P.; Gruzdev, D. A.; Levit, G. L. Eur. J. Org. Chem. 2012, 1471–1493. doi:10.1002/ejoc.201101489
Return to citation in text: [1] -
Duan, Y.; Chen, M.-W.; Chen, Q.-A.; Yu, C.-B.; Zhou, Y.-G. Org. Biomol. Chem. 2012, 10, 1235–1238. doi:10.1039/c1ob06777j
Return to citation in text: [1] -
Wang, Q.; Li, T.-R.; Lu, L.-Q.; Li, M.-M.; Zhang, K.; Xiao, W.-J. J. Am. Chem. Soc. 2016, 138, 8360–8363. doi:10.1021/jacs.6b04414
Return to citation in text: [1] -
Yang, Q.-Q.; Wang, Q.; An, J.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. Chem. – Eur. J. 2013, 19, 8401–8404. doi:10.1002/chem.201300988
Return to citation in text: [1] -
Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Rev. 2015, 115, 5301–5365. doi:10.1021/cr5006974
Return to citation in text: [1] -
Yang, Q.-Q.; Xiao, W.-J. Eur. J. Org. Chem. 2017, 233–236. doi:10.1002/ejoc.201601186
Return to citation in text: [1] -
Aher, R. D.; Suryavanshi, G. M.; Sudalai, A. J. Org. Chem. 2017, 82, 5940–5946. doi:10.1021/acs.joc.7b00439
Return to citation in text: [1] -
Pang, M.; Chang, H.; Feng, Z.; Zhang, J. Chin. J. Org. Chem. 2023, 43, 1271–1291. doi:10.6023/cjoc202210026
Return to citation in text: [1] -
Zhang, D.-Y.; Yu, C.-B.; Wang, M.-C.; Gao, K.; Zhou, Y.-G. Tetrahedron Lett. 2012, 53, 2556–2559. doi:10.1016/j.tetlet.2012.03.036
Return to citation in text: [1] -
Borrmann, R.; Knop, N.; Rueping, M. Chem. – Eur. J. 2017, 23, 798–801. doi:10.1002/chem.201605450
Return to citation in text: [1] -
Yang, Z.; Chen, F.; He, Y.; Yang, N.; Fan, Q.-H. Angew. Chem., Int. Ed. 2016, 55, 13863–13866. doi:10.1002/anie.201607890
Return to citation in text: [1] -
Liu, C.; Wang, M.; Xu, Y.; Li, Y.; Liu, Q. Angew. Chem., Int. Ed. 2022, 61, e202202814. doi:10.1002/anie.202202814
Return to citation in text: [1] -
Qin, L.; Deng, G.; Du, L.; Cui, B.; Wan, N.; Chen, Y. Tetrahedron Lett. 2022, 113, 154249. doi:10.1016/j.tetlet.2022.154249
Return to citation in text: [1] -
Zhao, W.; Zhang, Z.; Feng, X.; Yang, J.; Du, H. Org. Lett. 2020, 22, 5850–5854. doi:10.1021/acs.orglett.0c01930
Return to citation in text: [1] -
Xiao, Y.-C.; Wang, C.; Yao, Y.; Sun, J.; Chen, Y.-C. Angew. Chem., Int. Ed. 2011, 50, 10661–10664. doi:10.1002/anie.201105341
Return to citation in text: [1] -
Rueping, M.; Brinkmann, C.; Antonchick, A. P.; Atodiresei, I. Org. Lett. 2010, 12, 4604–4607. doi:10.1021/ol1019234
Return to citation in text: [1] -
Yang, K.; Lou, Y.; Wang, C.; Qi, L.-W.; Fang, T.; Zhang, F.; Xu, H.; Zhou, L.; Li, W.; Zhang, G.; Yu, P.; Song, Q. Angew. Chem., Int. Ed. 2020, 59, 3294–3299. doi:10.1002/anie.201913656
Return to citation in text: [1] -
Rueping, M.; Sugiono, E.; Azap, C.; Theissmann, T.; Bolte, M. Org. Lett. 2005, 7, 3781–3783. doi:10.1021/ol0515964
Return to citation in text: [1] -
Lackner, A. D.; Samant, A. V.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14090–14093. doi:10.1021/ja4082827
Return to citation in text: [1]
| 32. | Lackner, A. D.; Samant, A. V.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14090–14093. doi:10.1021/ja4082827 |
| 30. | Yang, K.; Lou, Y.; Wang, C.; Qi, L.-W.; Fang, T.; Zhang, F.; Xu, H.; Zhou, L.; Li, W.; Zhang, G.; Yu, P.; Song, Q. Angew. Chem., Int. Ed. 2020, 59, 3294–3299. doi:10.1002/anie.201913656 |
| 31. | Rueping, M.; Sugiono, E.; Azap, C.; Theissmann, T.; Bolte, M. Org. Lett. 2005, 7, 3781–3783. doi:10.1021/ol0515964 |
| 1. | Gupta, S. P. Lett. Drug Des. Discovery 2019, 16, 1089–1109. doi:10.2174/1570180816666190130154726 |
| 5. | Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Chem. – Eur. J. 2015, 21, 12836–12865. doi:10.1002/chem.201501475 |
| 6. | Meanwell, N. A. J. Med. Chem. 2011, 54, 2529–2591. doi:10.1021/jm1013693 |
| 26. | Qin, L.; Deng, G.; Du, L.; Cui, B.; Wan, N.; Chen, Y. Tetrahedron Lett. 2022, 113, 154249. doi:10.1016/j.tetlet.2022.154249 |
| 27. | Zhao, W.; Zhang, Z.; Feng, X.; Yang, J.; Du, H. Org. Lett. 2020, 22, 5850–5854. doi:10.1021/acs.orglett.0c01930 |
| 28. | Xiao, Y.-C.; Wang, C.; Yao, Y.; Sun, J.; Chen, Y.-C. Angew. Chem., Int. Ed. 2011, 50, 10661–10664. doi:10.1002/anie.201105341 |
| 4. | Ma, C.; Gao, P.-L.; Chu, J.; Wu, X.-P.; Wen, C.-W.; Kang, D.; Bai, J.-L.; Pei, X.-Y. Novel heterocyclic derivatives useful as shp2 inhibitors. International Patent Application WO2017/211303 A1, Dec 14, 2017. |
| 29. | Rueping, M.; Brinkmann, C.; Antonchick, A. P.; Atodiresei, I. Org. Lett. 2010, 12, 4604–4607. doi:10.1021/ol1019234 |
| 3. | Young, B. L.; Cooks, R. G.; Madden, M. C.; Bair, M.; Jia, J.; Aubry, A.-F.; Miller, S. A. J. Pharm. Biomed. Anal. 2007, 43, 1602–1608. doi:10.1016/j.jpba.2006.12.027 |
| 23. | Borrmann, R.; Knop, N.; Rueping, M. Chem. – Eur. J. 2017, 23, 798–801. doi:10.1002/chem.201605450 |
| 24. | Yang, Z.; Chen, F.; He, Y.; Yang, N.; Fan, Q.-H. Angew. Chem., Int. Ed. 2016, 55, 13863–13866. doi:10.1002/anie.201607890 |
| 2. | Zhang, L.; Zhang, X.; Cui, Y.; Yang, C. Synthesis 2021, 53, 3815–3826. doi:10.1055/a-1509-8624 |
| 25. | Liu, C.; Wang, M.; Xu, Y.; Li, Y.; Liu, Q. Angew. Chem., Int. Ed. 2022, 61, e202202814. doi:10.1002/anie.202202814 |
| 12. | Saito, K.; Akiyama, T. Angew. Chem., Int. Ed. 2016, 55, 3148–3152. doi:10.1002/anie.201510692 |
| 13. | Murray, J. I.; Flodén, N. J.; Bauer, A.; Fessner, N. D.; Dunklemann, D. L.; Bob‐Egbe, O.; Rzepa, H. S.; Bürgi, T.; Richardson, J.; Spivey, A. C. Angew. Chem., Int. Ed. 2017, 56, 5760–5764. doi:10.1002/anie.201700977 |
| 14. | Krasnov, V. P.; Gruzdev, D. A.; Levit, G. L. Eur. J. Org. Chem. 2012, 1471–1493. doi:10.1002/ejoc.201101489 |
| 16. | Wang, Q.; Li, T.-R.; Lu, L.-Q.; Li, M.-M.; Zhang, K.; Xiao, W.-J. J. Am. Chem. Soc. 2016, 138, 8360–8363. doi:10.1021/jacs.6b04414 |
| 17. | Yang, Q.-Q.; Wang, Q.; An, J.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. Chem. – Eur. J. 2013, 19, 8401–8404. doi:10.1002/chem.201300988 |
| 18. | Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Rev. 2015, 115, 5301–5365. doi:10.1021/cr5006974 |
| 19. | Yang, Q.-Q.; Xiao, W.-J. Eur. J. Org. Chem. 2017, 233–236. doi:10.1002/ejoc.201601186 |
| 20. | Aher, R. D.; Suryavanshi, G. M.; Sudalai, A. J. Org. Chem. 2017, 82, 5940–5946. doi:10.1021/acs.joc.7b00439 |
| 10. | Duan, Y.; Li, L.; Chen, M.-W.; Yu, C.-B.; Fan, H.-J.; Zhou, Y.-G. J. Am. Chem. Soc. 2014, 136, 7688–7700. doi:10.1021/ja502020b |
| 11. | Núñez-Rico, J. L.; Fernández-Pérez, H.; Vidal-Ferran, A. Green Chem. 2014, 16, 1153–1157. doi:10.1039/c3gc42132e |
| 21. | Pang, M.; Chang, H.; Feng, Z.; Zhang, J. Chin. J. Org. Chem. 2023, 43, 1271–1291. doi:10.6023/cjoc202210026 |
| 22. | Zhang, D.-Y.; Yu, C.-B.; Wang, M.-C.; Gao, K.; Zhou, Y.-G. Tetrahedron Lett. 2012, 53, 2556–2559. doi:10.1016/j.tetlet.2012.03.036 |
| 9. | Hua, T.-B.; Xiao, C.; Yang, Q.-Q.; Chen, J.-R. Chin. Chem. Lett. 2020, 31, 311–323. doi:10.1016/j.cclet.2019.07.015 |
| 7. | Famiglini, V.; La Regina, G.; Coluccia, A.; Masci, D.; Brancale, A.; Badia, R.; Riveira-Muñoz, E.; Esté, J. A.; Crespan, E.; Brambilla, A.; Maga, G.; Catalano, M.; Limatola, C.; Formica, F. R.; Cirilli, R.; Novellino, E.; Silvestri, R. J. Med. Chem. 2017, 60, 6528–6547. doi:10.1021/acs.jmedchem.6b01906 |
| 8. | Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Eur. J. Med. Chem. 2019, 183, 111691. doi:10.1016/j.ejmech.2019.111691 |
| 15. | Duan, Y.; Chen, M.-W.; Chen, Q.-A.; Yu, C.-B.; Zhou, Y.-G. Org. Biomol. Chem. 2012, 10, 1235–1238. doi:10.1039/c1ob06777j |
© 2024 Wang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.