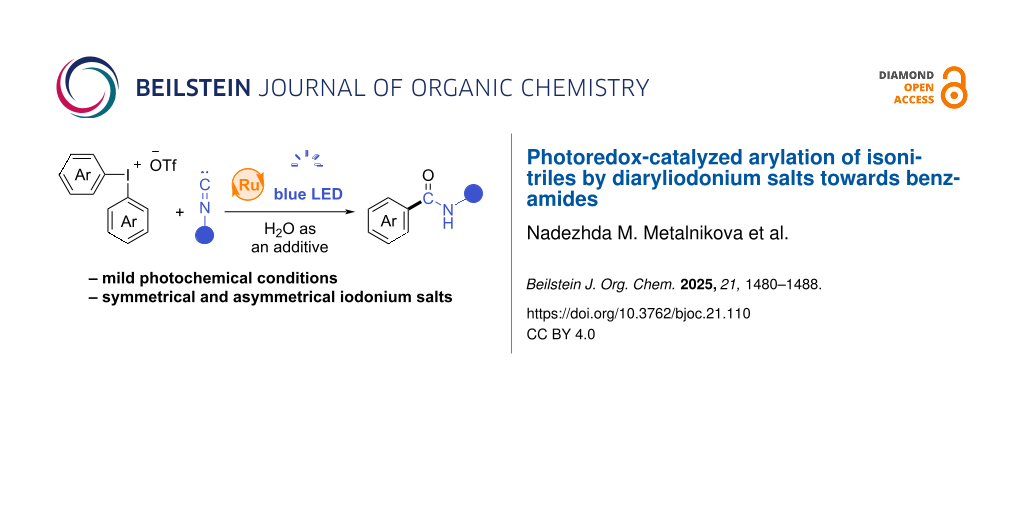
Abstract
The arylation of isonitriles by diaryliodonium salts under photoredox conditions has been proposed for the first time. The suggested procedure allows preparing a broad range of benzamides using both symmetric and unsymmetric diaryliodonium salts under mild conditions. A plausible mechanism for the reaction and the selectivity of aryl transfer (in case of unsymmetrical iodonium salts) were studied.

Graphical Abstract
Introduction
Amides represent a crucial and ubiquitous structural motif in essential biomolecules including proteins and peptides [1], as well as in a wide array of bioactive compounds. According to the DrugBank there are more than 250 approved drugs classified as amides [2]. Just recently, between February 2021 and June 2022, sixteen anticancer drugs containing an amide bond have been approved by the U.S. FDA [3]. Consequently, the preparation of amides has garnered significant attention within organic and medicinal chemistry. Commonly, amide bonds are formed via the reaction of carboxylic acids or their derivatives with appropriate amines (Scheme 1A) [4]. Although this conventional approach is effective and straightforward, it usually suffers from harsh conditions and low tolerance to sensitive functional groups. Due to these reasons, alternative routes toward the preparation of amides are still in great demand in modern synthetic chemistry [5].
![[1860-5397-21-110-i1]](/bjoc/content/inline/1860-5397-21-110-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The arylation of isonitriles was introduced nearly a decade ago as an alternative synthetic pathway for the preparation of benzamides (Scheme 1A) [6]. Over the years, the basic reaction has been modified to imply various metal-containing catalysts [7-18], metal-free transformations that employ heteroarenes under harsh conditions [19], or using diazonium salts as arylating agents [20,21]. However, diazonium salts are distinguished by their inherent instability complicating their use in such transformations. Moreover, among the various arylation strategies, photochemical methods remain relatively underexplored, with only a few examples reported [22,23]. These methods often face limitations such as the significant excess of isonitrile [22] or a restricted scope of aryl bromides (Scheme 1B) [23].
In contrast, diaryliodonium salts, representing stable, robust, and efficient arylating agents [24-30], have not been explored yet for the synthesis of benzamides from isonitriles as well as in multicomponent reactions with isonitriles in general. Only a few examples of photochemical cascade arylation–cyclizations of isonitriles with diaryliodonium salts have been published [31-33]. To bridge this gap, we propose a photoredox-mediated strategy for the synthesis of benzamides via the arylation of isonitriles with diaryliodonium salts under blue light irradiation (Scheme 1C).
Results and Discussion
We commenced our investigation by the optimization of the reaction conditions. During the preliminary experiments we tested different solvents and solvent-to-water ratios to establish initial conditions (Supporting Information File 1, 2.1 Solvent screening). Diphenyliodonium triflate (1a) and 1-isocyano-4-methylbenzene were used as model substrates. Taking into account the possibility of iodonium salt decomposition under irradiation we carried out an initial experiment without a catalyst and observed only traces of the benzamide 2aa (Table 1, entry 1). However, we detected almost half of the salt 1a remained in the reaction medium after 10 hours of reaction (Supporting Information File 1, Figure S3). Thus, we settled with the similar conditions to the published ones [31-33] introducing the photocatalyst [Ru(bpy)3](PF6)2, which successfully initiated the reaction under blue light irradiation and afforded benzamide 2aa in 36% yield (Table 1, entry 2). In that case less than 2% of the iodonium salt 1a remained in the reaction medium according to the 1H NMR spectrum of the crude mixture (Supporting Information File 1, Figure S4). Despite its higher reductive potential, fac-Ir(ppy)3 gave a lower yield of 26% for 2aa compared to [Ru(bpy)3](PF6)2 (Table 1, entry 3). Inspired by the first positive results, we tested various cyanoarene-based catalytic systems. Unfortunately, 4CzTPN, 4CzIPN, and 3DPAFIPN did not demonstrate increased efficiency, and the yields of the target product 2aa were slightly lower than for [Ru(bpy)3](PF6)2 (Table 1, entries 4–6). Thus, all further optimization studies were done using [Ru(bpy)3](PF6)2 as a catalyst.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-110-i5.svg?max-width=637&scale=1.0)
|
|||||
| # | Isonitrile/iodonium salt ratio | c (isonitrile) [M] | Catalystb | Base | Yield [%]c |
| variation of catalysts | |||||
| 1 | 1:1 | 0.1 | - | Na2CO3 | n.d. |
| 2 | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 36 (31)d |
| 3 | 1:1 | 0.1 | fac-Ir(ppy)3 | Na2CO3 | 26 |
| 4 | 1:1 | 0.1 | 4CzTPN | Na2CO3 | 20 |
| 5 | 1:1 | 0.1 | 4CzIPN | Na2CO3 | 30 |
| 6 | 1:1 | 0.1 | 3DPAFIPN | Na2CO3 | 30 |
| variation of bases | |||||
| 7 | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2HPO4 | 21 |
| 8 | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | NaH2PO4 | 24 |
| 9 | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | - | 22 |
| 10 | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Cs2CO3 | 30 |
| 11 | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | KOH | 11 |
| variation of reagents concentration | |||||
| 12 | 1:1 | 0.2 | [Ru(bpy)3](PF6)2 | Na2CO3 | 32 |
| 13 | 1:1 | 0.05 | [Ru(bpy)3](PF6)2 | Na2CO3 | 28 |
| variation of isonitrile/iodonium salt ratio | |||||
| 14 | 1:2 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 31 |
| 15 | 1:3 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 20 |
| 16 | 1.5:1 | 0.08 | [Ru(bpy)3](PF6)2 | Na2CO3 | 28 |
| 17 | 2:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 34 |
| 18 | 2:1 | 0.2 | [Ru(bpy)3](PF6)2 | Na2CO3 | 42 |
| 19 | 3:1 | 0.15 | [Ru(bpy)3](PF6)2 | Na2CO3 | 34 |
| 20 | 4:1 | 0.2 | [Ru(bpy)3](PF6)2 | Na2CO3 | 36 |
| 21 | 4:1 | 0.4 | [Ru(bpy)3](PF6)2 | Na2CO3 | 30 |
| variation of iodonium salts | |||||
| 22e | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 28 |
| 23f | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 37 |
| control experiments | |||||
| 24g | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | traces |
| 25h | 1:1 | 0.1 | [Ru(bpy)3](PF6)2 | Na2CO3 | 9 |
aReaction conditions: MeCN (1 mL), H2O (100 μL), irradiation by blue LED (465 nm, 20 W) for 10 h. bThe structures of photocatalysts are given in Supporting Information File 1, Figure S2. cDetermined by 1H NMR spectroscopy using 1,3,5-isopropylbenzene as an internal standard. dIsolated yield (for 0.2 mmol scale). e1a-BF4 was used instead of 1a. f1a-TsO was used instead of 1a. gIn the dark. hUnder air.
After, we moved to the screening of bases and their potential role in the arylation process. Firstly, the reaction without a base, or using weaker bases such as sodium phosphates, resulted in reduced yields of product 2aa (Table 1, entries 7–9) due to the acidic hydrolysis of the isonitrile to formamide [34]. Stronger bases such as Cs2CO3 (Table 1, entry 10) and KOH (Table 1, entry 11) led to diminished yields, reducing the product formation to 30% and 11%, respectively. In both cases we observed substantial decomposition of the iodonium salt affecting the yield of the desired product. We also performed the reaction with DIPEA as an organic base but the yield of 2aa was very low and we observed a range of byproducts, mainly amine addition instead of water (see Supporting Information File 1, 2.2 Preliminary and Additional Experiments, Figures S5 and S6).
Afterwards, we evaluated the other crucial parameters for photochemical reactions such as the reagents concentrations and ratio. Surprisingly, both dilution of the reaction mixture to 0.05 M and concentration to 0.2 M led to reduced yields (Table 1, entries 12 and 13) compared to the optimal concentration of 0.1 M. Furthermore, the yield of 2aa exhibited minimal dependence on the molar ratio of iodonium salt to isonitrile (Table 1, entries 14–21). In case of excess of the isonitrile we observed multiple addition and formation of oligomerized products which hindered the isolation of the product 2aa (Supporting Information File 1, 2.2 Preliminary and Additional Experiments, supplementary note 1).
It is known that the counteranion of an iodonium salt could be a crucial factor for the reactivity pattern. Thus, we tested iodonium salts with different anions. Reactions with diphenyliodonium tetrafluoroborate 1a-BF4 (Table 1, entry 22) and tosylate 1a-TsO (Table 1, entry 23) gave benzamide 2aa in 28% and 37% yield, respectively. Diphenyliodonium tosylate 1a-TsO afforded the product 2aa in a yield which was comparable to triflate 1a. However, we proceeded for further experiments with the triflate 1a since the triflates are generally more synthetically available.
Finally, control experiments without irradiation gave only traces of the benzamide 2aa showing no activation by the Ru complex at room temperature (Table 1, entry 24). An experiment conducted under an air atmosphere yielded only 9% of 2aa (Table 1, entry 25) indicating that the presence of atmospheric oxygen significantly inhibited the reaction.
Based on these findings and some additional experiments (see Supporting Information File 1, Table S1), the optimal conditions for further studies were established as the use of equimolar amounts of diaryliodonium salt and isonitrile, Na2CO3 as the base, and [Ru(bpy)3](PF6)2 as the photocatalyst, under an Ar atmosphere with irradiation by blue LED light (Table 1, entry 2).
With the optimized conditions in hands, a series of benzamides 2aa–je were synthesized using various symmetrical diaryliodonium salts 1a–k and isonitriles (Scheme 2). The analysis of reaction yields allowed us to establish the dependency from the electronic effects of substituents in the diaryliodonium salts. Diaryliodonium salts containing electron-deficient aryls afforded products 2 in higher yields compared to those bearing electron-donating groups (EDG). Specifically, the reaction with electron-withdrawing groups (EWG)-substituted iodonium salts produced benzamides 2bc–be, 2bg–bj in 18–67% yields. The benzamide 2bj was isolated in only 18% yield probably due to the low solubility of bis(3,5-ditrifluoromethyl)iodonium triflate (1j) in MeCN/H2O mixture. The highest yields were achieved in case of o-halo-substituted diaryliodonium salts providing 64% and 67% for amides 2bg and 2bh, respectively. In contrast, iodonium salts with electron-rich aryls afforded the corresponding benzamides 2aa, 2ba, 2bb, 2bf, and 2bk in significantly lower 19–36% yields.
![[1860-5397-21-110-i2]](/bjoc/content/inline/1860-5397-21-110-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction scope of iodonium salts 1 and isonitriles. aReaction conditions: isonitrile (0.2 mmol), iodonium salt 1 (0.2 mmol), Na2CO3 (0.34 mmol), [Ru(bpy)3](PF6)2 (0.004 mmol), MeCN (2 mL), H2O (200 μL), irradiation by blue LED (465 nm, 20 W) for 10 h. bYields in grey correspond to reactions with 2 equiv of iodonium salt 1 and 3.4 equiv of Na2CO3.
Scheme 2: Reaction scope of iodonium salts 1 and isonitriles. aReaction conditions: isonitrile (0.2 mmol), io...
Subsequently, various isonitriles were evaluated in the reaction with bis(4-bromophenyl)iodonium triflate (1e) under optimized conditions. The corresponding amides were successfully synthesized from both aliphatic and aromatic isonitriles, with yields reaching up to 61%. The highest yields were observed for benzamides 2be and 2ge in the reaction with sterically hindered aromatic isonitriles, such as 1-isocyano-2,4,6-trimethylbenzene and 1-isocyano-2,6-dimethylbenzene.
The use of 2 equivalents of iodonium salt 1 further improved the yields of certain amides (2bb, 2bj, 2de, 2ee, 2ge and 2he), as significant amounts of unreacted isonitrile remained when only 1 equivalent was employed.
The limited scope of iodonium salts for arylations usually arose from the poor range of synthetically accessible symmetrical salts compared to their unsymmetrical analogues. However, the selective transfer of one of the aryl groups is a main challenge for unsymmetrical iodonium salts. Therefore, we moved to these to test the selectivity of aryl transfer under the established conditions. Since iodonium salts are prone to repeat the selectivity pattern of nucleophilic substitution in photoredox processes [35-41], we evaluated iodonium salts with common dummy ligands such as 2,4,6-trimethoxyphenyl (TMP) [42,43], sterically hindered 2,4,6-triisopropylphenyl, and mesityl ligands. The highest selectivity was achieved using aryl(2,4,6-trimethoxyphenyl)iodonium triflates 1n–p yielding the desired amides 2ba, 2bo, and 2bp in 25–42% yield. Competing amide 2bn was not detected in the reaction mixture even by GC–MS analysis. In contrast, the mesityl-substituted salt 1l gave both competing products 2ba and 2bk in a ≈4:1 ratio, while the 2,4,6-triisopropylphenyl derivative 2bm yielded a mixture of products 2ba and 2bm in low yield (Scheme 3 and Supporting Information File 1, supplementary note 2).
![[1860-5397-21-110-i3]](/bjoc/content/inline/1860-5397-21-110-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Selectivity experiments and scope of unsymmetrical iodoniums salts. aReaction conditions: 2-isocyano-1,3-dimethylbenzene (0.2 mmol), iodonium salt 1 (0.2 mmol), Na2CO3 (0.34 mmol), [Ru(bpy)3](PF6)2 (0.004 mmol), MeCN (2 mL), H2O (200 μL), under irradiation by blue LED (465 nm, 20 W) for 10 h. bDetermined by 1H NMR spectroscopy after isolation as a mixture of amides or a single amide using 1,2-dibromoethane as an internal standard. cNot detected by GC–MS.
Scheme 3: Selectivity experiments and scope of unsymmetrical iodoniums salts. aReaction conditions: 2-isocyan...
Our experiments clearly demonstrated that electron-poor aryls gave better yields in case of both symmetrical and unsymmetrical iodonium salts. In general, such results cannot be associated with the stability of radical species, which does not significantly change for EWG- or EDG-substituted species except for extreme cases [44]. Additionally, such reactivity pattern cannot be explained only by steric factors since the yield dramatically dropped for o-methyl-substituted iodonium salt 1f compared to o-halo-substituted salts 1g and 1h, which provided the best yields in the scope. Moreover, the strong correlation of the yield with the electronic effects in aryl rings was clearly shown in the experiment with iodonium salt 1q (Scheme 3). We believe that the reason for the predominant transfer of the electron-poor ligands under the given conditions is due to a favorable formation of EWG-substituted aryl radicals from the iodonium cation, based on their reduction potentials and bond-dissociation energies calculated by Romanczyk and Kurek [45]. The reduction potential in SET reactions for iodonium salts with EWG-substituted aryls significantly differs from the ones with electron-rich aryls with 0.36 eV gap between (4-NO2C6H4)2I+ and (4-OMeC6H4)2I+ iodonium cations. If unsymmetric iodonium cations are considered where one of the aryls is phenyl and the other is a 4-substituted phenyl the bond-dissociation energy is 4.0 kcal/mol lower in case of (4-NO2C6H4) compared to (4-OMeC6H4) [45]. Therefore, despite the fact that literature data mostly suggest similar reactivity for aryl radicals with different substituents in the phenyl ring, the formation itself is more favorable for EWG-substituted radicals.
To gain a deeper understanding of the reactivity pattern in the current transformation, a reaction mechanism was proposed taking into the account the known data and control experiments (Scheme 4). Upon irradiation with blue light, the Ru(II) catalyst undergoes photoexcitation, followed by an oxidative single-electron transfer (SET) process with the iodonium salt, leading to the generation of an aryl radical, aryl iodide, and Ru(III). The formation of the aryl radical was corroborated through a trapping experiment utilizing TEMPO as a radical scavenger (Scheme 4 and Supporting Information File 1, 5. Control experiments, Figure S18). The resulting aryl radical is subsequently captured by an isonitrile molecule, forming an imidoyl radical intermediate X1. The intermediate X1 facilitates the reduction of the Ru(III) species back to Ru(II) thereby completing the photoredox cycle, with the formation of the cationic intermediate X2. We propose that bulky isonitriles effectively shield the radical or cationic centers in intermediates X1 or X2, thereby preventing multiple additions of isonitrile to give the highest yields for benzamides 2be and 2ge among the scope of isonitriles. In the final step of the reaction, the addition of a water molecule from the reaction medium to X2 occurs culminating in the formation of the final product 2 after deprotonation and tautomerization. The proposed mechanistic pathway is formally supported by conducting the reaction in the presence of NaOAc as a base, which resulted in the formation of the acetoxy derivative 3 attributable to the addition of acetate instead of water in the final step (Scheme 4 and Supporting Information File 1, 5. Control experiments, Figure S19). Additional control experiments excluded the possible arylation of formamide 4 by the iodonium salt (Supporting Information File 1, 5. Control experiments, Figure S20).
![[1860-5397-21-110-i4]](/bjoc/content/inline/1860-5397-21-110-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, a novel synthetic methodology for the preparation of benzamides from isonitriles and diaryliodonium salts has been proposed utilizing visible-light photoredox ruthenium-based catalysis. Both symmetrical and unsymmetrical diaryliodonium salts were evaluated and their reactivity was systematically analyzed in relation to the structural features of the iodonium salts and isonitriles. The study revealed that EWG-substituted diaryliodonium salts exhibited superior performance compared to EDG-substituted ones. Furthermore, the potential for selective transfer of a single aryl group from unsymmetrical diaryliodonium salts was demonstrated through the use of dummy ligands, such as 2,4,6-trimethoxyphenyl.
Supporting Information
| Supporting Information File 1: Experimental section, characterization data and control experiments. | ||
| Format: PDF | Size: 10.5 MB | Download |
Acknowledgements
Authors acknowledge the valuable advices from Yulia A. Vlasenko. TKN and МАК express their gratitude to the Center for Magnetic Resonance and the Center for Chemical Analysis and Materials Research at St Petersburg State University for providing access to analytical instrumentation essential for this research.
Data Availability Statement
Data generated and analyzed during this study is available from the corresponding author upon reasonable request.
References
-
Greenberg, A.; Breneman, C. M.; Liebman, J. F., Eds. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; John Wiley & Sons: New York, NY, USA, 2000.
Return to citation in text: [1] -
DrugBank Online, Accession number DBCAT000494. https://go.drugbank.com/categories/DBCAT000494 (accessed June 27, 2025).
Return to citation in text: [1] -
Asif, M.; Srivastava, R.; Fatima, A.; Shakeel, M.; Hassan, F.; Nasibullah, M. Russ. J. Bioorg. Chem. 2023, 49, 1165–1176. doi:10.1134/s1068162023060018
Return to citation in text: [1] -
Acosta‐Guzmán, P.; Ojeda‐Porras, A.; Gamba‐Sánchez, D. Adv. Synth. Catal. 2023, 365, 4359–4391. doi:10.1002/adsc.202301018
Return to citation in text: [1] -
Pattabiraman, V. R.; Bode, J. W. Nature 2011, 480, 471–479. doi:10.1038/nature10702
Return to citation in text: [1] -
Jiang, H.; Liu, B.; Li, Y.; Wang, A.; Huang, H. Org. Lett. 2011, 13, 1028–1031. doi:10.1021/ol103081y
Return to citation in text: [1] -
Huang, L.; Guo, H.; Pan, L.; Xie, C. Eur. J. Org. Chem. 2013, 6027–6031. doi:10.1002/ejoc.201300982
Return to citation in text: [1] -
Yavari, I.; Ghazanfarpour-Darjani, M.; Bayat, M. J. Tetrahedron Lett. 2014, 55, 4981–4982. doi:10.1016/j.tetlet.2014.05.032
Return to citation in text: [1] -
Khairnar, B. J.; Bhanage, B. M. Synthesis 2014, 46, 1236–1242. doi:10.1055/s-0033-1340733
Return to citation in text: [1] -
Sarkar, S.; Pal, R.; Roy, M.; Chatterjee, N.; Sarkar, S.; Sen, A. K. Tetrahedron Lett. 2015, 56, 623–626. doi:10.1016/j.tetlet.2014.12.060
Return to citation in text: [1] -
Li, Y.; Cao, J.; Zhu, Q.; Zhang, X.; Shi, G. Russ. J. Gen. Chem. 2016, 86, 668–671. doi:10.1134/s1070363216030257
Return to citation in text: [1] -
Lu, F.; Chen, Z.; Li, Z.; Wang, X.; Peng, X.; Li, C.; Fang, L.; Liu, D.; Gao, M.; Lei, A. Org. Lett. 2017, 19, 3954–3957. doi:10.1021/acs.orglett.7b01548
Return to citation in text: [1] -
Liu, J.-Q.; Shen, X.; Liu, Z.; Wang, X.-S. Org. Biomol. Chem. 2017, 15, 6314–6317. doi:10.1039/c7ob01449j
Return to citation in text: [1] -
Khalaj, M.; Taherkhani, M.; Mousavi-Safavi, S. M.; Akbari, J. Synlett 2018, 29, 94–98. doi:10.1055/s-0036-1588565
Return to citation in text: [1] -
Sharma, P.; Jain, N. Adv. Synth. Catal. 2018, 360, 1932–1937. doi:10.1002/adsc.201800107
Return to citation in text: [1] -
Yasaei, Z.; Mohammadpour, Z.; Shiri, M.; Tanbakouchian, Z.; Fazelzadeh, S. Front. Chem. (Lausanne, Switz.) 2019, 7, 433. doi:10.3389/fchem.2019.00433
Return to citation in text: [1] -
Li, Q.; Cai, Y.; Jin, H.; Liu, Y.; Zhou, B. Tetrahedron Lett. 2020, 61, 152605. doi:10.1016/j.tetlet.2020.152605
Return to citation in text: [1] -
Yan, Q.; Yuan, Q.-J.; Shatskiy, A.; Alvey, G. R.; Stepanova, E. V.; Liu, J.-Q.; Kärkäs, M. D.; Wang, X.-S. Org. Lett. 2024, 26, 3380–3385. doi:10.1021/acs.orglett.4c00872
Return to citation in text: [1] -
Zhou, Z.; Ji, H.; Li, Q.; Zhang, Q.; Li, D. Org. Biomol. Chem. 2021, 19, 2917–2922. doi:10.1039/d1ob00245g
Return to citation in text: [1] -
Basavanag, U. M. V.; Dos Santos, A.; El Kaim, L.; Gámez‐Montaño, R.; Grimaud, L. Angew. Chem., Int. Ed. 2013, 52, 7194–7197. doi:10.1002/anie.201302659
Return to citation in text: [1] -
Xia, Z.; Zhu, Q. Org. Lett. 2013, 15, 4110–4113. doi:10.1021/ol4017244
Return to citation in text: [1] -
Malacarne, M.; Protti, S.; Fagnoni, M. Adv. Synth. Catal. 2017, 359, 3826–3830. doi:10.1002/adsc.201700619
Return to citation in text: [1] [2] -
Cannalire, R.; Amato, J.; Summa, V.; Novellino, E.; Tron, G. C.; Giustiniano, M. J. Org. Chem. 2020, 85, 14077–14086. doi:10.1021/acs.joc.0c01946
Return to citation in text: [1] [2] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] -
Yusubov, M. S.; Maskaev, A. V.; Zhdankin, V. V. ARKIVOC 2011, No. i, 370–409. doi:10.3998/ark.5550190.0012.107
Return to citation in text: [1] -
Olofsson, B. Top. Curr. Chem. 2015, 373, 135–166. doi:10.1007/128_2015_661
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Mamgain, R.; Sakthivel, K.; Singh, F. V. Beilstein J. Org. Chem. 2024, 20, 2891–2920. doi:10.3762/bjoc.20.243
Return to citation in text: [1] -
Pan, C.; Wang, L.; Han, J. Chem. Rec. 2023, 23, e202300138. doi:10.1002/tcr.202300138
Return to citation in text: [1] -
Wang, R.; Jiang, H.; Cheng, Y.; Kadi, A. A.; Fun, H.-K.; Zhang, Y.; Yu, S. Synthesis 2014, 46, 2711–2726. doi:10.1055/s-0034-1379217
Return to citation in text: [1] [2] -
Jiang, H.; Cheng, Y.; Wang, R.; Zhang, Y.; Yu, S. Chem. Commun. 2014, 50, 6164–6167. doi:10.1039/c4cc01122h
Return to citation in text: [1] [2] -
Chen, J.-Y.; Wu, H.-Y.; Song, H.-Y.; Li, H.-X.; Jiang, J.; Yang, T.-B.; He, W.-M. J. Org. Chem. 2023, 88, 8360–8368. doi:10.1021/acs.joc.3c00380
Return to citation in text: [1] [2] -
Sung, K.; Chen, C.-C. Tetrahedron Lett. 2001, 42, 4845–4848. doi:10.1016/s0040-4039(01)00863-2
Return to citation in text: [1] -
Samanta, R. K.; Meher, P.; Murarka, S. J. Org. Chem. 2022, 87, 10947–10957. doi:10.1021/acs.joc.2c01234
Return to citation in text: [1] -
Chen, Z.; Liu, N.-W.; Bolte, M.; Ren, H.; Manolikakes, G. Green Chem. 2018, 20, 3059–3070. doi:10.1039/c8gc00838h
Return to citation in text: [1] -
Liu, N.-W.; Liang, S.; Manolikakes, G. Adv. Synth. Catal. 2017, 359, 1308–1319. doi:10.1002/adsc.201601341
Return to citation in text: [1] -
Senapati, S.; Parida, S. K.; Karandikar, S. S.; Murarka, S. Org. Lett. 2023, 25, 7900–7905. doi:10.1021/acs.orglett.3c03146
Return to citation in text: [1] -
Meher, P.; Samanta, R. K.; Manna, S.; Murarka, S. Chem. Commun. 2023, 59, 6092–6095. doi:10.1039/d3cc00605k
Return to citation in text: [1] -
Thombare, K. R.; Parida, S. K.; Meher, P.; Murarka, S. Chem. Commun. 2024, 60, 13907–13910. doi:10.1039/d4cc05086j
Return to citation in text: [1] -
Galicia, J.; McDonald, N. R.; Bennett, C. W.; He, J.; Glossbrenner, M. D.; Romero, E. A. Chem. Commun. 2024, 60, 6929–6932. doi:10.1039/d4cc01259c
Return to citation in text: [1] -
Stuart, D. R. Chem. – Eur. J. 2017, 23, 15852–15863. doi:10.1002/chem.201702732
Return to citation in text: [1] -
Sandtorv, A. H.; Stuart, D. R. Angew. Chem., Int. Ed. 2016, 55, 15812–15815. doi:10.1002/anie.201610086
Return to citation in text: [1] -
Parsaee, F.; Senarathna, M. C.; Kannangara, P. B.; Alexander, S. N.; Arche, P. D. E.; Welin, E. R. Nat. Rev. Chem. 2021, 5, 486–499. doi:10.1038/s41570-021-00284-3
Return to citation in text: [1] -
Romańczyk, P. P.; Kurek, S. S. Electrochim. Acta 2020, 351, 136404. doi:10.1016/j.electacta.2020.136404
Return to citation in text: [1] [2]
| 44. | Parsaee, F.; Senarathna, M. C.; Kannangara, P. B.; Alexander, S. N.; Arche, P. D. E.; Welin, E. R. Nat. Rev. Chem. 2021, 5, 486–499. doi:10.1038/s41570-021-00284-3 |
| 35. | Samanta, R. K.; Meher, P.; Murarka, S. J. Org. Chem. 2022, 87, 10947–10957. doi:10.1021/acs.joc.2c01234 |
| 36. | Chen, Z.; Liu, N.-W.; Bolte, M.; Ren, H.; Manolikakes, G. Green Chem. 2018, 20, 3059–3070. doi:10.1039/c8gc00838h |
| 37. | Liu, N.-W.; Liang, S.; Manolikakes, G. Adv. Synth. Catal. 2017, 359, 1308–1319. doi:10.1002/adsc.201601341 |
| 38. | Senapati, S.; Parida, S. K.; Karandikar, S. S.; Murarka, S. Org. Lett. 2023, 25, 7900–7905. doi:10.1021/acs.orglett.3c03146 |
| 39. | Meher, P.; Samanta, R. K.; Manna, S.; Murarka, S. Chem. Commun. 2023, 59, 6092–6095. doi:10.1039/d3cc00605k |
| 40. | Thombare, K. R.; Parida, S. K.; Meher, P.; Murarka, S. Chem. Commun. 2024, 60, 13907–13910. doi:10.1039/d4cc05086j |
| 41. | Galicia, J.; McDonald, N. R.; Bennett, C. W.; He, J.; Glossbrenner, M. D.; Romero, E. A. Chem. Commun. 2024, 60, 6929–6932. doi:10.1039/d4cc01259c |
| 42. | Stuart, D. R. Chem. – Eur. J. 2017, 23, 15852–15863. doi:10.1002/chem.201702732 |
| 43. | Sandtorv, A. H.; Stuart, D. R. Angew. Chem., Int. Ed. 2016, 55, 15812–15815. doi:10.1002/anie.201610086 |
| 1. | Greenberg, A.; Breneman, C. M.; Liebman, J. F., Eds. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; John Wiley & Sons: New York, NY, USA, 2000. |
| 5. | Pattabiraman, V. R.; Bode, J. W. Nature 2011, 480, 471–479. doi:10.1038/nature10702 |
| 31. | Wang, R.; Jiang, H.; Cheng, Y.; Kadi, A. A.; Fun, H.-K.; Zhang, Y.; Yu, S. Synthesis 2014, 46, 2711–2726. doi:10.1055/s-0034-1379217 |
| 32. | Jiang, H.; Cheng, Y.; Wang, R.; Zhang, Y.; Yu, S. Chem. Commun. 2014, 50, 6164–6167. doi:10.1039/c4cc01122h |
| 33. | Chen, J.-Y.; Wu, H.-Y.; Song, H.-Y.; Li, H.-X.; Jiang, J.; Yang, T.-B.; He, W.-M. J. Org. Chem. 2023, 88, 8360–8368. doi:10.1021/acs.joc.3c00380 |
| 4. | Acosta‐Guzmán, P.; Ojeda‐Porras, A.; Gamba‐Sánchez, D. Adv. Synth. Catal. 2023, 365, 4359–4391. doi:10.1002/adsc.202301018 |
| 34. | Sung, K.; Chen, C.-C. Tetrahedron Lett. 2001, 42, 4845–4848. doi:10.1016/s0040-4039(01)00863-2 |
| 3. | Asif, M.; Srivastava, R.; Fatima, A.; Shakeel, M.; Hassan, F.; Nasibullah, M. Russ. J. Bioorg. Chem. 2023, 49, 1165–1176. doi:10.1134/s1068162023060018 |
| 24. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 25. | Yusubov, M. S.; Maskaev, A. V.; Zhdankin, V. V. ARKIVOC 2011, No. i, 370–409. doi:10.3998/ark.5550190.0012.107 |
| 26. | Olofsson, B. Top. Curr. Chem. 2015, 373, 135–166. doi:10.1007/128_2015_661 |
| 27. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 28. | Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 29. | Mamgain, R.; Sakthivel, K.; Singh, F. V. Beilstein J. Org. Chem. 2024, 20, 2891–2920. doi:10.3762/bjoc.20.243 |
| 30. | Pan, C.; Wang, L.; Han, J. Chem. Rec. 2023, 23, e202300138. doi:10.1002/tcr.202300138 |
| 2. | DrugBank Online, Accession number DBCAT000494. https://go.drugbank.com/categories/DBCAT000494 (accessed June 27, 2025). |
| 31. | Wang, R.; Jiang, H.; Cheng, Y.; Kadi, A. A.; Fun, H.-K.; Zhang, Y.; Yu, S. Synthesis 2014, 46, 2711–2726. doi:10.1055/s-0034-1379217 |
| 32. | Jiang, H.; Cheng, Y.; Wang, R.; Zhang, Y.; Yu, S. Chem. Commun. 2014, 50, 6164–6167. doi:10.1039/c4cc01122h |
| 33. | Chen, J.-Y.; Wu, H.-Y.; Song, H.-Y.; Li, H.-X.; Jiang, J.; Yang, T.-B.; He, W.-M. J. Org. Chem. 2023, 88, 8360–8368. doi:10.1021/acs.joc.3c00380 |
| 20. | Basavanag, U. M. V.; Dos Santos, A.; El Kaim, L.; Gámez‐Montaño, R.; Grimaud, L. Angew. Chem., Int. Ed. 2013, 52, 7194–7197. doi:10.1002/anie.201302659 |
| 21. | Xia, Z.; Zhu, Q. Org. Lett. 2013, 15, 4110–4113. doi:10.1021/ol4017244 |
| 22. | Malacarne, M.; Protti, S.; Fagnoni, M. Adv. Synth. Catal. 2017, 359, 3826–3830. doi:10.1002/adsc.201700619 |
| 19. | Zhou, Z.; Ji, H.; Li, Q.; Zhang, Q.; Li, D. Org. Biomol. Chem. 2021, 19, 2917–2922. doi:10.1039/d1ob00245g |
| 23. | Cannalire, R.; Amato, J.; Summa, V.; Novellino, E.; Tron, G. C.; Giustiniano, M. J. Org. Chem. 2020, 85, 14077–14086. doi:10.1021/acs.joc.0c01946 |
| 7. | Huang, L.; Guo, H.; Pan, L.; Xie, C. Eur. J. Org. Chem. 2013, 6027–6031. doi:10.1002/ejoc.201300982 |
| 8. | Yavari, I.; Ghazanfarpour-Darjani, M.; Bayat, M. J. Tetrahedron Lett. 2014, 55, 4981–4982. doi:10.1016/j.tetlet.2014.05.032 |
| 9. | Khairnar, B. J.; Bhanage, B. M. Synthesis 2014, 46, 1236–1242. doi:10.1055/s-0033-1340733 |
| 10. | Sarkar, S.; Pal, R.; Roy, M.; Chatterjee, N.; Sarkar, S.; Sen, A. K. Tetrahedron Lett. 2015, 56, 623–626. doi:10.1016/j.tetlet.2014.12.060 |
| 11. | Li, Y.; Cao, J.; Zhu, Q.; Zhang, X.; Shi, G. Russ. J. Gen. Chem. 2016, 86, 668–671. doi:10.1134/s1070363216030257 |
| 12. | Lu, F.; Chen, Z.; Li, Z.; Wang, X.; Peng, X.; Li, C.; Fang, L.; Liu, D.; Gao, M.; Lei, A. Org. Lett. 2017, 19, 3954–3957. doi:10.1021/acs.orglett.7b01548 |
| 13. | Liu, J.-Q.; Shen, X.; Liu, Z.; Wang, X.-S. Org. Biomol. Chem. 2017, 15, 6314–6317. doi:10.1039/c7ob01449j |
| 14. | Khalaj, M.; Taherkhani, M.; Mousavi-Safavi, S. M.; Akbari, J. Synlett 2018, 29, 94–98. doi:10.1055/s-0036-1588565 |
| 15. | Sharma, P.; Jain, N. Adv. Synth. Catal. 2018, 360, 1932–1937. doi:10.1002/adsc.201800107 |
| 16. | Yasaei, Z.; Mohammadpour, Z.; Shiri, M.; Tanbakouchian, Z.; Fazelzadeh, S. Front. Chem. (Lausanne, Switz.) 2019, 7, 433. doi:10.3389/fchem.2019.00433 |
| 17. | Li, Q.; Cai, Y.; Jin, H.; Liu, Y.; Zhou, B. Tetrahedron Lett. 2020, 61, 152605. doi:10.1016/j.tetlet.2020.152605 |
| 18. | Yan, Q.; Yuan, Q.-J.; Shatskiy, A.; Alvey, G. R.; Stepanova, E. V.; Liu, J.-Q.; Kärkäs, M. D.; Wang, X.-S. Org. Lett. 2024, 26, 3380–3385. doi:10.1021/acs.orglett.4c00872 |
| 45. | Romańczyk, P. P.; Kurek, S. S. Electrochim. Acta 2020, 351, 136404. doi:10.1016/j.electacta.2020.136404 |
| 6. | Jiang, H.; Liu, B.; Li, Y.; Wang, A.; Huang, H. Org. Lett. 2011, 13, 1028–1031. doi:10.1021/ol103081y |
| 22. | Malacarne, M.; Protti, S.; Fagnoni, M. Adv. Synth. Catal. 2017, 359, 3826–3830. doi:10.1002/adsc.201700619 |
| 23. | Cannalire, R.; Amato, J.; Summa, V.; Novellino, E.; Tron, G. C.; Giustiniano, M. J. Org. Chem. 2020, 85, 14077–14086. doi:10.1021/acs.joc.0c01946 |
| 45. | Romańczyk, P. P.; Kurek, S. S. Electrochim. Acta 2020, 351, 136404. doi:10.1016/j.electacta.2020.136404 |
© 2025 Metalnikova et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.