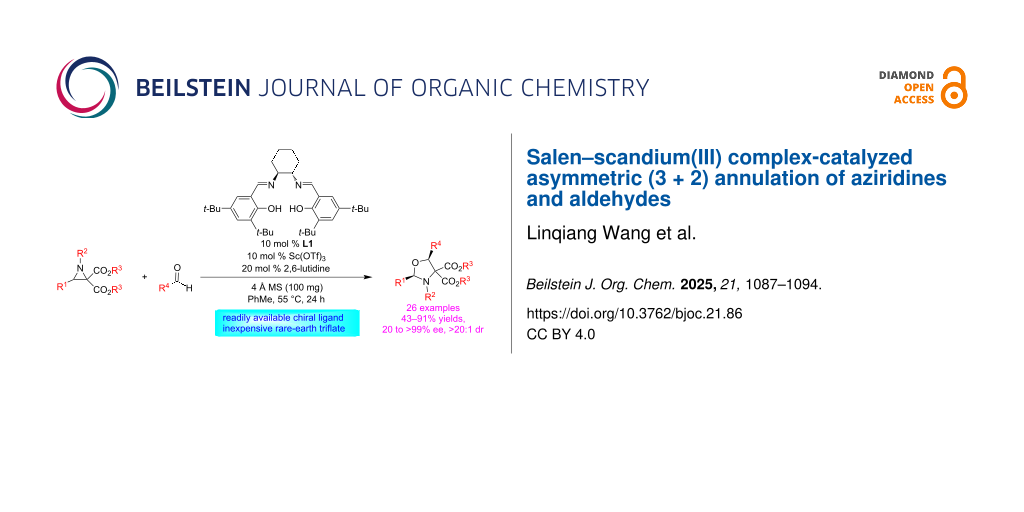
Abstract
Oxazolidine is one of the crucial structural moieties of biologically active compounds. A salen–scandium triflate complex-catalyzed asymmetric (3 + 2) annulation of dialkyl 1-sulfonylaziridine-2,2-dicarboxylates and aldehydes generated optically active functionalized oxazolidine derivatives in moderate to good yields and good to excellent enantioselectivities and high diastereoselectivities. A reasonable reaction mechanism was proposed and rationalized the experimental results.

Graphical Abstract
Introduction
Oxazolidine derivatives are an important class of nitrogen and oxygen-containing five-membered saturated heterocycles. They are not only useful building blocks for the synthesis of biologically active compounds [1], for example, side-chain precursor of paclitaxel [2], but also widely exist in some pharmaceuticals [3,4], such as antibacterial agents of Gram-positive organisms [5] and the FDA-approved antibiotic linezolid [6] (Figure 1). Both chiral oxazolidines [7,8] and oxazolidinones [9,10] have been utilized as chiral auxiliary groups in many asymmetric organic transformations.
![[1860-5397-21-86-1]](/bjoc/content/figures/1860-5397-21-86-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Oxazolidine-containing bioactive compounds.
Figure 1: Oxazolidine-containing bioactive compounds.
Oxazolidine derivatives have been prepared mainly from condensation of vicinal amino alcohols and aldehydes [11] and through [2 + 3] cycloaddition of azomethine ylides and carbonyl dipolarophiles [12]. Recently, the [2 + 3] annulation of aldehydes and donor–acceptor dialkyl 2-aryl-1-sulfonylaziridine-2,2-dicarboxylates, which generate azomethine ylides, has been developed for the synthesis of racemic and optically active functionalized cis-2,5-diaryloxazolidine derivatives [13-16]. Racemic cis-2,5-diaryloxazolidine derivatives were prepared under the catalysis of zinc triflate or nickel diperchlorate (Scheme 1a) [13,14]. Later, highly enantiomeric cis-2,5-diaryloxazolidine derivatives were obtained under asymmetric catalysis with a Ni(II)–bisoxazoline complex [15] and a Nd(OTf)3/N,N'-dioxide/LiNTf2 delay catalytic system [16], respectively (Scheme 1b and c). However, further exploration for convenient asymmetric catalytic synthetic methods is still in demand because the former require dineopentyl aziridine-2,2-dicarboxylates to realize high enantioselectivity, while the synthesis of the chiral ligand N,N'-dioxide requires multiple steps. Herein, we present a convenient highly diastereo- and enantioselective synthesis of dialkyl 2,5-diaryl-1-sulfonyloxazolidine-2,2-dicarboxylates from aldehydes and dialkyl 2-aryl-1-sulfonylaziridine-2,2-dicarboxylates under the catalysis of the readily available salen–Sc(OTf)3 complex (Scheme 1d). The salen ligand can be prepared in one step from enantiopure cyclohexane-1,2-diamines and substituted salicylaldehydes.
![[1860-5397-21-86-i1]](/bjoc/content/inline/1860-5397-21-86-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Asymmetric catalytic synthetic methods of oxazolidine derivatives.
Scheme 1: Asymmetric catalytic synthetic methods of oxazolidine derivatives.
Results and Discussion
The reaction of diethyl 3-phenyl-1-(4-toluenesulfonyl)aziridine-2,2-dicarboxylate (1a) and benzaldehyde (2a) was first selected as a model reaction to optimize the reaction conditions (Table 1). When aziridine 1a (0.2 mmol) and aldehyde 2a (0.3 mmol) in dried toluene (1 mL) were stirred at 10 °C for 35 h in the presence of commercially available Sc(OTf)3 (0.01 mmol, 5 mol %), only a trace amount of product 3aa was observed (Table 1, entry 1). When chiral ligand L1 (0.01 mmol) was added, a trace amount of product 3aa was obtained with 17% ee (Table 1, entry 2). The enantioselectivity increased to 32% when 2,6-lutidine (0.02 mmol) was added (Table 1, entry 3). When pre-dried Sc(OTf)3 was used instead of commercially available one, the reaction generated the desired product 3aa in 39% yield with 70% ee and >20:1 diastereoselectivity (Table 1, entry 4). The product 3aa was identified as diethyl (2R,5S)-2,5-diphenyl-3-tosyloxazolidine-4,4-dicarboxylate on the basis of comparison with reported NMR and specific rotation data [16]. The enantiomeric excess increased to 96% when the amounts of Sc(OTf)3 and L1 were increased to 10 mol %, but the yield remained moderate (38%) (Table 1, entry 5). Other rare-earth salts, including Y(OTf)3, La(OTf)3, Sm(OTf)3, Tb(OTf)3, Er(OTf)3, and Lu(OTf)3, were also screened, but no better results were observed (Table 1, entries 6–11). In addition, different ligands L2–L4 were evaluated (Table 1, entries 12–14). The enantiomer of product 3aa was obtained in 28% yield with 31% ee and >20:1 diastereomeric ratio in the presence of ligand L2 (Table 1, entry 12). However, ligands L3 and L4 were completely inactive. Thus, further optimizations were performed with 10 mol % of Sc(OTf)3 and L1. The reaction was conducted at different temperatures for 12 h for saving time. The yield was improved from 21% to 29% to 34% with similar enantio- and diastereoselectivities at 25 °C, 45 °C, and 55 °C, respectively, but the enantioselectivity decreased slightly at 55 °C (Table 1, entries 15–17). Further extending the reaction time to 48 h at 55 °C, resulted in an increased yield of 60%, but the enantioselectivity decreased to 90% (Table 1, entry 18). Solvent screening indicated that toluene was the best choice (Table 1, entries 18–21). Because Sc(OTf)3 is moisture sensitive and the reaction is obviously impacted by the quality of Sc(OTf)3, Sc(OTf)3 was strictly dried at 220 °C for 2 h under an oil pump vacuum and used in further optimizations in toluene with or without simultaneously dried molecular sieves. When the reaction was conducted at 55 °C for 24 h, product 3aa was obtained in 61% yield and 98%ee in the presences of MS and in 49% yield and 97% ee without MS. Finally, the optimal reaction conditions were selected as: Sc(OTf)3 (0.02 mmol) and 4 Å MS (100 mg) were dried at 220 °C for 2 h under oil pump vacuum. 2,6-Lutidine (0.04 mmol), L1 (0.02 mmol), and dry toluene (1 mL) were added and the mixture was stirred at 55 °C for 3 h. To the solution was added a solution of aziridine 1a (0.2 mmol) and aldehyde 2a (0.3 mmol) in dry toluene (1 mL) and the resulting mixture was stirred at 55 °C for 24 h.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-86-i5.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | Ligand | RE(OTf)3 | Base | Solv | Temp [°C] | Time [h] | Yield [%]b |
ee [%]
cisc |
dr
cis/transd |
| 1e | – | Sc(OTf)3 | – | PhMe | 10 | 35 | trace | – | – |
| 2e | L1 | Sc(OTf)3 | – | PhMe | 10 | 35 | trace | 17 | – |
| 3e | L1 | Sc(OTf)3 | lutidine | PhMe | 10 | 35 | trace | 32 | – |
| 4f | L1 | Sc(OTf)3 | lutidine | PhMe | 10 | 35 | 39 | 70 | >20:1 |
| 5 | L1 | Sc(OTf)3 | lutidine | PhMe | 10 | 35 | 38 | 96 | >20:1 |
| 6 | L1 | Y(OTf)3 | lutidine | PhMe | 10 | 35 | 33 | 4 | 60:40 |
| 7 | L1 | La(OTf)3 | lutidine | PhMe | 10 | 35 | 22 | 50 | 79:21 |
| 8 | L1 | Sm(OTf)3 | lutidine | PhMe | 10 | 35 | 29 | 0 | >19:1 |
| 9 | L1 | Tb(OTf)3 | lutidine | PhMe | 10 | 35 | 49 | 0 | >20:1 |
| 10 | L1 | Er(OTf)3 | lutidine | PhMe | 10 | 35 | 31 | 0 | >20:1 |
| 11 | L1 | Lu(OTf)3 | lutidine | PhMe | 10 | 35 | 46 | −6 | >20:1 |
| 12 | L2 | Sc(OTf)3 | lutidine | PhMe | 10 | 35 | 28 | −31 | >20:1 |
| 13 | L3 | Sc(OTf)3 | lutidine | PhMe | 10 | 35 | N.R. | – | – |
| 14 | L4 | Sc(OTf)3 | lutidine | PhMe | 10 | 35 | N.R. | – | – |
| 15 | L1 | Sc(OTf)3 | lutidine | PhMe | 25 | 12 | 21 | 96 | >20:1 |
| 16 | L1 | Sc(OTf)3 | lutidine | PhMe | 45 | 12 | 29 | 96 | >20:1 |
| 17 | L1 | Sc(OTf)3 | lutidine | PhMe | 55 | 12 | 34 | 94 | >20:1 |
| 18 | L1 | Sc(OTf)3 | lutidine | PhMe | 55 | 48 | 60 | 90 | >20:1 |
| 19 | L1 | Sc(OTf)3 | lutidine | THF | 55 | 24 | 14 | 84 | >20:1 |
| 20 | L1 | Sc(OTf)3 | lutidine | MeCN | 55 | 24 | 19 | 36 | >20:1 |
| 21 | L1 | Sc(OTf)3 | lutidine | DCE | 55 | 24 | 16 | 88 | >20:1 |
| 22g | L1 | Sc(OTf)3 | lutidine | PhMe | 55 | 24 | 61 | 98 | >20:1 |
| 23h | L1 | Sc(OTf)3 | lutidine | PhMe | 55 | 24 | 49 | 97 | >20:1 |
aUnless otherwise noted, the reactions were performed as follows: Sc(OTf)3 (9.8 mg, 0.02 mmol) dried in a vacuum drying oven at 106 °C for 12 h, 2,6-lutidine (lutidine for short in Table 1) (4.7 μL, 0.04 mmol), and L1 (10.9 mg, 0.02 mmol) in solvent (1 mL) were stirred for 3 h. To the solution was added a solution of aziridine 1a (0.2 mmol) and aldehyde 2a (0.3 mmol) in solvent (1 mL). bIsolated by flash basic Al2O3 column chromatography. cDetermined by chiral HPLC analysis AD-H, hexane/iPrOH 70:30, flow rate = 0.8 mL/min, λ = 210 nm. dDetermined by 1H NMR spectroscopy. eUndried Sc(OTf)3 was used. fUndried Sc(OTf)3 (4.9 mg, 0.01mmol), L1 (5.5 mg, 0.01 mmol). gSc(OTf)3 and 4 Å molecular sieves (MS) (100 mg) were dried at 220 °C for 2 h under oil pump vacuum and used. hSc(OTf)3 was dried at 220 °C for 2 h under oil pump vacuum.
With the optimal reaction conditions in hand, the scope and generality of both aziridines 1 and aldehydes 2 were investigated (Scheme 2). Different aldehydes 2 were first reacted with diethyl 3-phenyl-1-(4-toluene)sulfonylaziridine-2,2-dicarboxylate (1a), affording cis-oxazolidines 3aa–aj in 43–84% yields and 37–98% ee. Obvious influences of electronic and steric effects on both the yields and enantioselectivities were observed. Aromatic aldehydes 2e and 2f with electron-withdrawing para-substituents generally gave the desired products 3ae and 3af in higher yields and enantioselectivities than aldehydes with electron-donating substituents (2b–d). Sterically congested 3-chlorobenzaldehyde (2g) and 2,6-dichlorobenzaldehyde (2h) produced the desired products 3ag and 3ah in lower yields and enantioselectivities than 4-chlorobenzaldehyde (2e). Multisubstituted 3,4,6-trimethoxybenzaldehyde (2i) also delivered the corresponding product 3ai in good yield (74%) and enantioselectivity (80% ee). However, heteroaromatic furan-2-carbaldehyde (2j) yielded the expected product 3aj in moderate yield (65%) and low enantioselectivity (41% ee). The reactions of different aldehydes 2 and diethyl 1-methanesulfonyl-3-phenylaziridine-2,2-dicarboxylate (1b) were performed, generating the corresponding oxazolidines 3ba–bo in 52–91% yields and 35–99% ee. Similar influence tendencies of electronic effect on the yield and stereoselectivity were observed as those in the reactions involving 1a, but the enantioselectivities were generally higher than those in reactions involving 1a. Electron-rich benzaldehydes 2c and 2d and sterically bulky benzaldehydes 2i and 2k generated the desired products 3bc, 3bd, 3bi, and 3bk in relatively low yields and enantioselectivities. The strongly electron-deficient 4-nitrobenzaldehyde (2l) showed the highest enantioselectivity, affording the desired product 3bl in 63% yield and >99% ee. Polycyclic fused naphthalene-1-carbaldehyde (2m) and heteroaromatic thiophen-2-carbaldehyde (2n) also smoothly underwent reaction, affording the expected products 3bm in 70% yield and 98% ee and 3bn in 82% yield and 96% ee, respectively. The reaction could be extended to 4-bromocinnamaldehyde (2o), giving the desired product 3bo in 91% yield and 50% ee. Furthermore, other diethyl 2-phenylaziridine-2,2-dicarboxylates 1c and 1d with different 1-arenesulfonyl groups were attempted, giving rise to the corresponding products 3ca and 3da in 87% yield with 88% ee and in 82% yield with 20% ee. The results indicate that aziridine 1d with an electron-deficient 1-(4-chlorophenyl)sulfonyl group presented low enantioselectivity. Next, aziridines 1e and 1f with methyl and isopropyl carboxylates also showed excellent enantioselectivities as the corresponding aziridine 1b with ethyl carboxylate groups. Finally, diethyl 3-(4-bromophenyl)-1-tosylaziridine-2,2-dicarboxylate (1g) was attempted as well, delivering the expected product 3ga in 70% yield and 38% ee, illustrating that the 3-aryl group of aziridines 1 also has an important effect on the enantioselectivity in the (2 + 3) annulation. Compared with previously reported methods, our current method is more convenient and uses readily available components.
![[1860-5397-21-86-i2]](/bjoc/content/inline/1860-5397-21-86-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of aziridines and aldehydes.
Scheme 2: Scope of aziridines and aldehydes.
On the basis of the experimental results and a previous report [16], a possible reaction mechanism is presented in Scheme 3. The catalyst (Cat) is first generated from salen L1 and Sc(OTf)3. Aziridines 1 coordinate to the Sc ion in the catalyst with their two carboxylate groups followed by a ring opening of the aziridine ring, forming azomethine ylide intermediates A. The intermediates A further undergo a [2 + 3] annulation (or cycloaddition) with aldehydes 2 through endo transition state TS to generate intermediates B, which release products 3 with regeneration of the catalyst for the next catalytic cycle. The electron-deficient aromatic aldehydes exhibit excellent stereoselectivity due to the π-stacking interaction between their aryl group and the electron-rich malonate group. Similar π-stacking interaction-controlled stereoselectivities were observed in our previous studies [17-20].
![[1860-5397-21-86-i3]](/bjoc/content/inline/1860-5397-21-86-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
To show the practicality of the reaction, a gram-scale reaction of 1b and 2f was carried out, giving the desired product 3bf in 87% yield (1.372 g) and 90% ee (Scheme 4). The yield was slightly higher than that in the micromolar reaction, but the enantioselectivity obviously was lower compared to the smaller scale reaction.
![[1860-5397-21-86-i4]](/bjoc/content/inline/1860-5397-21-86-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
A convenient and efficient synthetic method for the synthesis of optically active (2R,5S)-oxazolidines has been developed with aldehydes and dialkyl 3-aryl-1-sulfonylaziridine-2,2-dicarboxylates as starting materials. The method uses readily available salen as chiral ligand, which coordinates with scandium triflate to generate a salen–Sc complex acting as efficient catalyst. The catalytic asymmetric (3 + 2) annulation of dialkyl 3-aryl-1-sulfonylaziridine-2,2-dicarboxylates and aldehydes generated optically active functionalized oxazolidine derivatives in moderate to good yields and good to excellent enantioselectivities and high diastereoselectivities. The stereocontrol was rationalized in the proposed reaction mechanism.
Experimental
Unless otherwise noted, all materials were purchased from commercial suppliers. Toluene was refluxed over sodium with benzophenone as an indicator and freshly distilled prior to use. Column chromatography was performed on silica gel (normal phase, 200–300 mesh) from Anhui Liangchen Silicon Material Co., Ltd. or basic aluminum oxide (pH 9–10) from Shanghai Titan Technology Co., Ltd. Petroleum ether (PE, 60–90 °C fraction) and ethyl acetate (EA) were used as eluent. Reactions were monitored by thin-layer chromatography (TLC) on GF254 silica gel plates (0.2 mm) from Anhui Liangchen Silicon Material Co., Ltd. The plates were visualized by UV light. 1H NMR (400 MHz) and 13C NMR (101 MHz) spectra were recorded on a Bruker 400 NMR spectrometer (Billerica, MA, USA), usually with TMS as an internal standard for 1H NMR and the centered peak of CDCl3 as an internal standard (77.16) for 13C NMR in CDCl3 solution. The chemical shifts (δ) were reported in parts per million (ppm) relative to tetramethylsilane (TMS). Melting points were obtained on a melting point apparatus. HRMS measurements were carried out on an LC/MSD TOF mass spectrometer. Specific rotations were measured on an Anton Paar MCP500 polarimeter (Singapore) and are reported as follows: ![[Graphic 2]](/bjoc/content/inline/1860-5397-21-86-i6.svg?max-width=637&scale=1.18182) (c in g/100 mL, solvent). The enantiomeric excesses were determined using chiral HPLC analysis using an Agilent 1260 LC instrument (Santa Clara, CA, USA) with Daicel Chiralcel AD-H column (Hyderabad, India) with a mixture of isopropyl alcohol and hexane as eluents. All liquid aldehydes were washed with saturated aqueous sodium bicarbonate solution, dried over sodium sulfate, and freshly distilled prior to use. All solid aldehydes were used after recrystallization from petroleum ether (PE, 60–90 °C fraction) or a mixture of ethanol and water. All dialkyl 3-aryl-1-sulfonylaziridine-2,2-dicarboxylates 1 were synthesized by previously reported procedure [16].
(c in g/100 mL, solvent). The enantiomeric excesses were determined using chiral HPLC analysis using an Agilent 1260 LC instrument (Santa Clara, CA, USA) with Daicel Chiralcel AD-H column (Hyderabad, India) with a mixture of isopropyl alcohol and hexane as eluents. All liquid aldehydes were washed with saturated aqueous sodium bicarbonate solution, dried over sodium sulfate, and freshly distilled prior to use. All solid aldehydes were used after recrystallization from petroleum ether (PE, 60–90 °C fraction) or a mixture of ethanol and water. All dialkyl 3-aryl-1-sulfonylaziridine-2,2-dicarboxylates 1 were synthesized by previously reported procedure [16].
General procedure for the synthesis of ethyl 2-(oxazol-2-yl)alkanoates 3
Sc(OTf)3 (9.8 mg, 0.02 mmol) was added in a dried 10 mL reaction tube, then it was heated to 220 °C and dried at 220 °C for 2 h under oil pump vacuum. After gradually cooling to room temperature, the chiral salen ligand L1 (10.9 mg, 0.02 mmol), 4 Å MS (100 mg), 2,6-lutidine (4.7 μL, 0.04 mmol), and dry toluene (1 mL) were added. The resulting mixture was stirred at 55 °C for 3 h under a nitrogen atmosphere. A solution of aziridine 1 (0.2 mmol) and aldehyde (0.3 mmol) in 1 mL of dry toluene was added into the mixture with a syringe and the mixture was stirred at 55 °C for 24 h. After gradually cooling to room temperature and removal of the solvent under reduced pressure, the crude residue was purified by basic aluminum oxide column chromatography with petroleum ether/ethyl acetate 1:10 to 3:7 (v/v) as eluent to afford the pure product 3. The enantiomeric excess was determined using chiral HPLC analysis with a mixture of iPrOH and hexane as eluent.
Supporting Information
| Supporting Information File 1: Analytic data and copies of 1H and 13C NMR spectra of compounds 1 and 3, copies of HRMS spectra of unknown compound 3 and copies of HLPC profiles of compounds 3. | ||
| Format: PDF | Size: 9.7 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Morales‐Monarca, G. H.; Gnecco, D.; Terán, J. L. Eur. J. Org. Chem. 2022, e202200267. doi:10.1002/ejoc.202200267
Return to citation in text: [1] -
Jing, Y.-r.; Zhou, W.; Li, W.-l.; Zhao, L.-x.; Wang, Y.-f. Bioorg. Med. Chem. 2014, 22, 194–203. doi:10.1016/j.bmc.2013.11.037
Return to citation in text: [1] -
Wang, G. Anti-Infect. Agents Med. Chem. 2008, 7, 32–49. doi:10.2174/187152108783329771
Return to citation in text: [1] -
Branco-Junior, J. F.; Teixeira, D. R. C.; Pereira, M. C.; Pitta, I. R.; Galdino-Pitta, M. R. Curr. Bioact. Compd. 2017, 13, 292–304. doi:10.2174/1573407213666161214162149
Return to citation in text: [1] -
Yu, L.; Zhou, W.; Wang, Z. Bioorg. Med. Chem. Lett. 2011, 21, 1541–1544. doi:10.1016/j.bmcl.2010.12.097
Return to citation in text: [1] -
Mao, E.; MacMillan, D. W. C. J. Am. Chem. Soc. 2023, 145, 2787–2793. doi:10.1021/jacs.2c13396
Return to citation in text: [1] -
Agami, C.; Couty, F. Eur. J. Org. Chem. 2004, 677–685. doi:10.1002/ejoc.200300452
Return to citation in text: [1] -
Wolf, C.; Xu, H. Chem. Commun. 2011, 47, 3339–3350. doi:10.1039/c0cc04629a
Return to citation in text: [1] -
Ager, D. J.; Prakash, I.; Schaad, D. R. Aldrichimica Acta 1997, 30, 3–12.
Return to citation in text: [1] -
Mourad, A. K.; Czekelius, C. Top. Heterocycl. Chem. 2020, 55, 113–156. doi:10.1007/7081_2020_36
Return to citation in text: [1] -
Xu, X.; Wei, H.; Feng, H. Chem. Heterocycl. Compd. 2020, 56, 464–466. doi:10.1007/s10593-020-02681-w
Return to citation in text: [1] -
Meyer, A. G.; Ryan, J. H. Molecules 2016, 21, 935. doi:10.3390/molecules21080935
Return to citation in text: [1] -
Jiang, Z.; Wang, J.; Lu, P.; Wang, Y. Tetrahedron 2011, 67, 9609–9617. doi:10.1016/j.tet.2011.09.085
Return to citation in text: [1] [2] -
Wu, X.; Li, L.; Zhang, J. Chem. Commun. 2011, 47, 7824–7826. doi:10.1039/c1cc12189h
Return to citation in text: [1] [2] -
Wu, X. X.; Zhou, W.; Wu, H.-H.; Zhang, J. L. Chem. Commun. 2017, 53, 5661–5664. doi:10.1039/c7cc02906c
Return to citation in text: [1] [2] -
Liao, Y.; Liu, X.; Zhang, Y.; Xu, Y.; Xia, Y.; Lin, L.; Feng, X. Chem. Sci. 2016, 7, 3775–3779. doi:10.1039/c5sc04151a
Return to citation in text: [1] [2] [3] [4] [5] -
Xu, J. Molecules 2024, 29, 1454. doi:10.3390/molecules29071454
Return to citation in text: [1] -
Ma, L.; Jiao, P.; Zhang, Q.; Xu, J. Tetrahedron: Asymmetry 2005, 16, 3718–3734. doi:10.1016/j.tetasy.2005.09.025
Return to citation in text: [1] -
Ma, L.; Du, D.-M.; Xu, J. J. Org. Chem. 2005, 70, 10155–10158. doi:10.1021/jo051765y
Return to citation in text: [1] -
Wang, C.; Chen, N.; Yang, Z.; Xu, J. Molecules 2024, 29, 1963. doi:10.3390/molecules29091963
Return to citation in text: [1]
| 16. | Liao, Y.; Liu, X.; Zhang, Y.; Xu, Y.; Xia, Y.; Lin, L.; Feng, X. Chem. Sci. 2016, 7, 3775–3779. doi:10.1039/c5sc04151a |
| 1. | Morales‐Monarca, G. H.; Gnecco, D.; Terán, J. L. Eur. J. Org. Chem. 2022, e202200267. doi:10.1002/ejoc.202200267 |
| 6. | Mao, E.; MacMillan, D. W. C. J. Am. Chem. Soc. 2023, 145, 2787–2793. doi:10.1021/jacs.2c13396 |
| 16. | Liao, Y.; Liu, X.; Zhang, Y.; Xu, Y.; Xia, Y.; Lin, L.; Feng, X. Chem. Sci. 2016, 7, 3775–3779. doi:10.1039/c5sc04151a |
| 5. | Yu, L.; Zhou, W.; Wang, Z. Bioorg. Med. Chem. Lett. 2011, 21, 1541–1544. doi:10.1016/j.bmcl.2010.12.097 |
| 17. | Xu, J. Molecules 2024, 29, 1454. doi:10.3390/molecules29071454 |
| 18. | Ma, L.; Jiao, P.; Zhang, Q.; Xu, J. Tetrahedron: Asymmetry 2005, 16, 3718–3734. doi:10.1016/j.tetasy.2005.09.025 |
| 19. | Ma, L.; Du, D.-M.; Xu, J. J. Org. Chem. 2005, 70, 10155–10158. doi:10.1021/jo051765y |
| 20. | Wang, C.; Chen, N.; Yang, Z.; Xu, J. Molecules 2024, 29, 1963. doi:10.3390/molecules29091963 |
| 3. | Wang, G. Anti-Infect. Agents Med. Chem. 2008, 7, 32–49. doi:10.2174/187152108783329771 |
| 4. | Branco-Junior, J. F.; Teixeira, D. R. C.; Pereira, M. C.; Pitta, I. R.; Galdino-Pitta, M. R. Curr. Bioact. Compd. 2017, 13, 292–304. doi:10.2174/1573407213666161214162149 |
| 16. | Liao, Y.; Liu, X.; Zhang, Y.; Xu, Y.; Xia, Y.; Lin, L.; Feng, X. Chem. Sci. 2016, 7, 3775–3779. doi:10.1039/c5sc04151a |
| 2. | Jing, Y.-r.; Zhou, W.; Li, W.-l.; Zhao, L.-x.; Wang, Y.-f. Bioorg. Med. Chem. 2014, 22, 194–203. doi:10.1016/j.bmc.2013.11.037 |
| 16. | Liao, Y.; Liu, X.; Zhang, Y.; Xu, Y.; Xia, Y.; Lin, L.; Feng, X. Chem. Sci. 2016, 7, 3775–3779. doi:10.1039/c5sc04151a |
| 12. | Meyer, A. G.; Ryan, J. H. Molecules 2016, 21, 935. doi:10.3390/molecules21080935 |
| 13. | Jiang, Z.; Wang, J.; Lu, P.; Wang, Y. Tetrahedron 2011, 67, 9609–9617. doi:10.1016/j.tet.2011.09.085 |
| 14. | Wu, X.; Li, L.; Zhang, J. Chem. Commun. 2011, 47, 7824–7826. doi:10.1039/c1cc12189h |
| 11. | Xu, X.; Wei, H.; Feng, H. Chem. Heterocycl. Compd. 2020, 56, 464–466. doi:10.1007/s10593-020-02681-w |
| 15. | Wu, X. X.; Zhou, W.; Wu, H.-H.; Zhang, J. L. Chem. Commun. 2017, 53, 5661–5664. doi:10.1039/c7cc02906c |
| 9. | Ager, D. J.; Prakash, I.; Schaad, D. R. Aldrichimica Acta 1997, 30, 3–12. |
| 10. | Mourad, A. K.; Czekelius, C. Top. Heterocycl. Chem. 2020, 55, 113–156. doi:10.1007/7081_2020_36 |
| 7. | Agami, C.; Couty, F. Eur. J. Org. Chem. 2004, 677–685. doi:10.1002/ejoc.200300452 |
| 8. | Wolf, C.; Xu, H. Chem. Commun. 2011, 47, 3339–3350. doi:10.1039/c0cc04629a |
| 13. | Jiang, Z.; Wang, J.; Lu, P.; Wang, Y. Tetrahedron 2011, 67, 9609–9617. doi:10.1016/j.tet.2011.09.085 |
| 14. | Wu, X.; Li, L.; Zhang, J. Chem. Commun. 2011, 47, 7824–7826. doi:10.1039/c1cc12189h |
| 15. | Wu, X. X.; Zhou, W.; Wu, H.-H.; Zhang, J. L. Chem. Commun. 2017, 53, 5661–5664. doi:10.1039/c7cc02906c |
| 16. | Liao, Y.; Liu, X.; Zhang, Y.; Xu, Y.; Xia, Y.; Lin, L.; Feng, X. Chem. Sci. 2016, 7, 3775–3779. doi:10.1039/c5sc04151a |
© 2025 Wang and Xu; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.