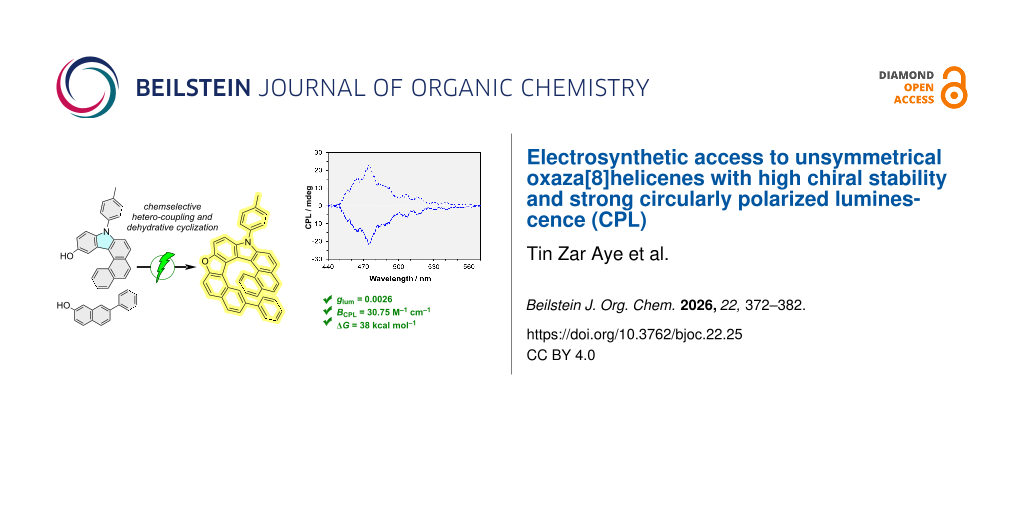
Abstract
Heterohelicenes are compelling chiral π-conjugated scaffolds for optoelectronic and chiral-photonic technologies because their helical frameworks and doped heteroatoms endow them with various photophysical, chiroptical, and electronic merits. However, unsymmetrical heterohelicenes remain rare, as their synthesis is often hindered by chemoselectivity and regioselective control. Here, we exploit the differential redox potentials of two coupling partners as a key player to achieve a chemo- and regioselective electrosynthetic access to a new family of unsymmetrical oxaza[8]helicenes. A controlled anodic sequence enables selective oxidative hetero-coupling followed by dehydrative cyclization, furnishing the extended [8]helical scaffold efficiently under mild, oxidant-free conditions. Structural analyses show retained aromaticity, increased helical distortion, and higher configurational stability (≈38 kcal/mol) relative to their oxaza[7]helicene analogues (<25 kcal/mol). After chiral HPLC separation, the enantiomers display mirror-image CD and strong solution CPL, with |glum| up to 2.6 × 10−3 and fluorescence brightness up to 30.75 M−1 cm−1.

Graphical Abstract
Introduction
Chirality is a pervasive feature of natural and artificial systems, and chiral small molecules continue to underpin advances in chemistry and materials science [1,2]. Among them, helicenes – ortho-condensed polycyclic aromatic hydrocarbons (PAHs) built from angularly annulated rings – occupy a distinctive niche because their non-planar, screw-shaped architectures generate inherent, configurational chirality [3-5]. This helicity originates from intramolecular steric congestion and stabilizing π–π interactions between terminal rings, yielding stable enantiomeric conformers with pronounced optical activity. The combination of rigid helical topology and tunable electronic structure has propelled helicenes into diverse applications, spanning chiral photonics [6-9], organic electronics [10,11], molecular machines [12], molecular recognition [13], and bioimaging [14]. Yet, most unsubstituted carbo[n]helicenes (n ≥ 5) often display modest fluorescence quantum yields, constraining their utility in emissive technologies [15]. Incorporation of heteroatoms to form hetero[n]helicenes provides an effective means to modulate frontier orbitals, intermolecular interactions, and excited-state dynamics, frequently enhancing fluorescence efficiency and circularly polarized luminescence (CPL) [16-22]. Consequently, heterohelicenes have emerged as attractive platforms for optoelectronic devices, 3D displays, security inks, and information-storage materials, where both helicity and emission characteristics must be precisely controlled [23-26].
A central design element in helicene chemistry is helical extension. Increasing the number of ortho-fused rings amplifies π-conjugation, structural rigidity, and chiral stability, typically strengthening chiroptical responses [27-29]; however, it also escalates synthetic difficulty due to heightened strain and more demanding regio- and chemoselective construction. While [7]helicenes have been extensively explored, their [8]helicene counterparts remain comparatively underdeveloped [30], despite the appealing prospect of higher barriers to enantiomerization and richer optoelectronic behavior [31]. In 2021, Yorimitsu and co-workers disclosed a series of symmetric dihetero[8]helicenes I–IV that exhibited intriguing chiroptical properties utilizing the characteristic transformations of the organosulfur functionality [32,33]. In 2024, Badani, Karnik, and co-workers accessed one member of this class, 7,12-dioxa[8]helicene I, through a sequence featuring photochemical E–Z isomerization, electrocyclization, and oxidative aromatization [34]. In parallel, Liu and co-workers introduced the π-extended azabora[8]helicene V with exceptional chiroptical signals and high brightness, emphasizing the promise of this class of chiral molecules (Scheme 1A) [35].
![[1860-5397-22-25-i1]](/bjoc/content/inline/1860-5397-22-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Recent examples of hetero[8]helicenes: (A) symmetric hetero[8]helicenes; (B) unsymmetrical hetero[8]helicenes; (C) short-step electrosynthetic access to new unsymmetrical oxaza[8]helicenes.
Scheme 1: Recent examples of hetero[8]helicenes: (A) symmetric hetero[8]helicenes; (B) unsymmetrical hetero[8...
Despite these advances, unsymmetrical hetero[8]helicenes – where heteroatoms occupy non-equivalent positions along the helical rim – are far rarer. Their scarcity primarily reflects the formidable challenge of controlling chemo- and regioselectivity during ring annulation and heteroatom introduction, which often necessitates multistep synthetic strategies [36-38]. To the best of our knowledge, only four examples have been reported so far [39-42]. Recent reports suggest that breaking symmetry can further amplify CPL responses and enable finer electronic tuning [39]. Voituriez, Marinetti and co-workers developed a phosphorus-embedded [8]helicene VI [40], Crassous and co-workers prepared azabora[8]helicene VII via cycloborylation of a pyridine-substituted carbo[6]helicene [41], while Xu, Wang and colleagues reported a multistep route to azabora[8]helicene VIII (Scheme 1B) [42]. Collectively, these studies highlight both the synthetic bottleneck and the untapped potential of unsymmetrical hetero[8]helicenes.
Inspired by the superior selectivity and sustainability of organic electrosynthesis as an eco-friendly alternative to conventional oxidative methods [43-47], we leveraged our electrochemical approaches [48-52], and redesigned the synthons to access a new unsymmetrical hetero[8]helicene (Scheme 1C). This strategy delivers the target scaffold in only three steps from commercially available substrates and exploits the differential oxidation potentials of the two partners to enforce chemoselective cross-annulation. To the best of our knowledge, this represents the shortest route reported to any unsymmetrical hetero[8]helicene. We then investigated the structural, photophysical and chiroptical properties of these new oxaza[8]helicenes and benchmarked their behavior against their corresponding oxaza[7]helicene analogues.
Results and Discussion
Electrosynthesis of unsymmetrical oxaza[8]helicenes
Building on Zhang’s facile acid-mediated carbazole synthesis [53], in which aniline derivatives react with p-benzoquinone to afford 3-hydroxycarbazoles [54], we employed a closely related substrate. Specifically, N-(p-tolyl)phenanthren-3-amine (2) – prepared from 3-bromophenanthrene (1) via Buchwald–Hartwig amination with p-toluidine under Pd catalysis [55] – was subjected to phosphoric acid-mediated annulation with p-benzoquinone to give the hydroxycarbazole derivative 3 through a tandem Michael addition/ring-closure sequence. After rapid optimization of key parameters (see Supporting Information File 1), we developed a one-pot electrochemical annulation between 3 and β-naphthol derivative 4. Using n-Bu4NPF6 as the electrolyte in CH2Cl2 at room temperature, this protocol furnished oxaza[8]helicenes 5 in good-to-moderate yields with >75% Faradaic efficiency, and no homo-coupling products were detected under the optimized conditions (Scheme 2).
![[1860-5397-22-25-i2]](/bjoc/content/inline/1860-5397-22-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Short-step synthesis of unsymmetrical oxaza[8]helicenes 5.
Scheme 2: Short-step synthesis of unsymmetrical oxaza[8]helicenes 5.
Based on our previous reports [48,56], DFT calculations, and cyclic voltammetry (CV) analyses (Figure 1), anodic single-electron transfer (SET) is expected to occur first from 3, generating the electrophilic radical cation [3]·+ as 3 (Eox = 0.735 V vs Fc/Fc+ in CH2Cl2) is oxidized more readily than the 2-naphthol partners (Eox of 4a = 1.081 V and Eox of 4b = 1.286 V vs Fc/Fc+ in CH2Cl2). The radical cation [3]·+ then undergoes rapid deprotonation to form a neutral radical intermediate (Int-I) with high spin density at the reactive site, enabling regioselective intermolecular coupling with 4. While a Scholl-type coupling-first scenario cannot be ruled out, the computed acidity of [3]·+ (pKa ≈ −5.2) together with the more spin-density localization in Int-I supports a deprotonation-first, neutral-radical pathway, consistent with related electrochemical arenol activations reported by Waldvogel and co-workers [46]. Subsequent intramolecular dehydrative cyclization furnishes the desired oxaza[8]helicenes 5. The oxidation-potential gap between 3 and 4 and the reactivity of Int-I thus provides a handle to control chemo- and regioselectivity.
![[1860-5397-22-25-1]](/bjoc/content/figures/1860-5397-22-25-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: A plausible reaction mechanism: cyclic voltammetry (CV) analyses of hydroxycarbazole derivative 3 and 2-naphthol derivatives 4; DFT-based calculations of the pKa of [3]·+ radical cation and spin density of neutral radical intermediate Int-I (optimized at the UB3LYP/6-31G+(d,p) level of theory with IEPCM model as solvation of DCM. Grimme’s dispersion with the original D3 damping function was applied as empirical dispersion correction to the optimized structures).
Figure 1: A plausible reaction mechanism: cyclic voltammetry (CV) analyses of hydroxycarbazole derivative 3 a...
Structural properties of oxaza[8]helicenes
Aromaticity
We evaluated the aromaticity of the oxaza[8]helicenes 5a and 5b using nucleus-independent chemical shifts (NICS), as proposed by Schleyer and co-workers [57,58]. As shown in Figure 2A, the terminal rings exhibit higher aromatic character and a progressive increase in aromaticity upon helical elongation compared with the corresponding oxaza[7] analogues (see Supporting Information File 1) [49], as evidenced by NICS(1)zz values of −26.97 and −21.17 for rings E and H in 5a, and −28.86 and −31.74 for rings E′ and H′ in 5b. This enhancement can be attributed to magnetic coupling between the face-to-face terminal rings, in line with the Johnson–Bovey model, given the inter-ring distances of approximately 3.75–3.79 Å [59]. On the other hand, the aromaticity of the pyrrole rings (A and A′) and benzene rings G and G′ in 5a and 5b decreases, which can be attributed to the increased deviation from planarity, particularly in the more extended helicenes, consistent with previous studies [60].
![[1860-5397-22-25-2]](/bjoc/content/figures/1860-5397-22-25-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Aromaticity of oxaza[8]helicenes: (A) NICS(0)zz and NICS(1)zz values of 5a and 5b calculated at MN15/6-311G(2d,p)/SMD=chloroform level of theory; (B) ACID plots calculated at the B3LYP/6-311G(d,p) level of theory (isosurface value: 0.05).
Figure 2: Aromaticity of oxaza[8]helicenes: (A) NICS(0)zz and NICS(1)zz values of 5a and 5b calculated at MN1...
To gain further insight, we performed anisotropy of the induced current density (AICD) calculations for 5a and 5b at the B3LYP/6-311G(d,p) level in the gas phase [61]. The resulting plots show a clockwise diatropic current along the fused heterocycles and benzene rings, in agreement with the ring-current patterns reported for other helicene scaffolds [27] (Figure 2B).
Enantiomerization barriers of oxaza[7]helicenes
To investigate the enantiomerization (P/M) barriers of oxaza[8]helicenes 5a and 5b, we performed DFT calculations to locate the transition states with the highest Gibbs free energies. In both cases, the transition states correspond to conformations in which the terminal rings adopt a face-to-face arrangement along the helical axis (Figure 3). The calculated enantiomerization barriers for 5a and 5b are 38.24 and 38.10 kcal mol−1, respectively (Figure 3A and 3B), highlighting the pronounced effect of π-extension on the rigidity of the helical backbone. In contrast, the corresponding oxaza[7]helicenes 6a and 6b exhibit significantly lower barriers of 21.05 and 24.91 kcal mol−1, respectively (Figure 3C and 3D), which leads to rapid enantiomerization within a few hours at room temperature and severely limits their applicability in chiroptical devices despite their favorable CD and CPL properties (vide infra). By comparison, the markedly higher (P/M) enantiomerization barriers of 5a and 5b translate into excellent configurational robustness, as demonstrated by the absence of detectable enantiomerization when solutions of (M)-5a were heated at 130 °C for 2.5 h.
![[1860-5397-22-25-3]](/bjoc/content/figures/1860-5397-22-25-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (P/M) Enantiomerization process of 5a (A), 5b (B), 6a (C), and 6b (D); relative Gibbs free energies were calculated in (kcal mol−1) at the MN15/6-31G(2d,p)/SMD=chloroform level of theory.
Figure 3: (P/M) Enantiomerization process of 5a (A), 5b (B), 6a (C), and 6b (D); relative Gibbs free energies...
Optical properties of oxaza[8]helicenes
Photophysical features
The absorption and emission spectra of oxaza[8]helicenes 5a and 5b in chloroform (1 × 10−5 M) were recorded and compared with those of the corresponding oxaza[7]helicenes 6a and 6b (Figure 4A and 4B). As expected, extension of the helical π-systems in 5 leads to enhanced conjugation relative to 6, manifested in red-shifted absorption and emission bands. In chloroform, 5a shows a pronounced higher absorption peak at 426 nm (ε = 8.23 × 104 M−1 cm−1) with an optical indirect bandgap (Eg) of 2.78 eV. Similarly, 5b, 6a, and 6b exhibit their higher absorption peaks at 433 nm (ε = 5.59 × 104 M−1 cm−1), 407 nm (ε = 6.43 × 104 M−1 cm−1), and 414 nm (ε = 9.02 × 104 M−1 cm−1), with corresponding optical indirect bandgaps Eg of 2.70, 2.93, and 2.85 eV, respectively (see Supporting Information File 1). The photoluminescence (PL) spectra in chloroform display emission maxima at 459 and 468 nm for 5a and 5b, compared to 439 and 447 nm for 6a and 6b.
![[1860-5397-22-25-4]](/bjoc/content/figures/1860-5397-22-25-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Photophysical characters of oxaza[n]helicenes: (A) and (B) UV–vis absorption and PL spectra; (C) Frontier Kohn–Sham molecular orbitals (HOMO and LUMO) optimized in the lowest energy excited state (S1) and TD-DFT calculated transitions at MN15/6-311G(2d,p)/SMD=chloroform level of theory.
Figure 4: Photophysical characters of oxaza[n]helicenes: (A) and (B) UV–vis absorption and PL spectra; (C) Fr...
To gain insight into the electronic transitions, we performed time-dependent DFT (TD-DFT) calculations for all oxaza[7]helicenes 6 and oxaza[8]helicenes 5 after geometry optimization at the S1 minimum (see Supporting Information File 1 and Supporting Information File 2) [62,63]. The convergence of these structures was confirmed by frequency analysis, which revealed no imaginary frequencies. The frontier orbitals are non-degenerate, and the S1 → S0 transition is dominated by the LUMO → HOMO contribution. The oscillator strength of the S1 → S0 transition decreases with helical elongation (Figure 4C), in line with the observed trends in emission efficiency. Fluorescence quantum yields (Φf, in chloroform, 1 × 10−3 M) for 5a and 5b were 25.1% and 22.6%, slightly lower than those of 6a (40.5%) and 6b (38.9%). This difference can be rationalized by their radiative rate constants (kf). The calculated kf,calcd values are 0.174, 0.159, 0.243, and 0.194 ns−1 for 5a, 5b, 6a, and 6b, respectively (see Supporting Information File 1). According to Φf = kf /( kf + knr), where knr is the non-radiative rate constant, the balance between these decay pathways accounts for the observed variation in quantum yields [64].
Chiroptical features
The higher enantiomerization barriers of (P/M)-5a and (P/M)-5b enabled complete separation of their enantiomers by HPLC using a Daicel Chiralpak IA column (see Supporting Information File 1). Despite the rapid enantiomerization of (P/M)-6a and (P/M)-6b, we were able to separate the two enantiomers at lower temperature and samples were stored at −20 °C before their chiroptical responses were evaluated. The optical purities of (P/M)-6a and (P/M)-6b measured samples were confirmed to be >97% ee, confirming the reliability of our results. However, this low enantiomerization barriers of oxaza[7]helicenes 6a,b hinders their practical applications. The CD spectra of optically pure 5a,b and 6a,b were recorded (Figure 5A), and compared with reported analogous oxaza[7]helicenes [49], and spectra obtained from TD-DFT to assign their absolute configurations [27]. The absolute configurations in the first and second fractions of the chiral HPLC analysis were assigned as the (P)- and (M)-enantiomers, respectively, for all 5a,b and 6a,b. As expected, the increase in helical length (n) from 7 to 8, 5a and 5b exhibited more red-shifted maximum |gabs| values at around 350 nm, whereas 6a and 6b showed values around 290–300 nm for both enantiomers (Figure 5A). High |gabs| values have also been reported for π-extended helical nanographenes featuring aza[7]helicene subunits [65].
![[1860-5397-22-25-5]](/bjoc/content/figures/1860-5397-22-25-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Chiroptical properties of oxaza[n]helicenes: (A) CD spectra measured in chloroform (1 × 10−5 M); CPL spectra measured in chloroform (1 × 10−3 M). Solid lines for (M)-configuration and dashed lines for (P)-configuration of 5a (black), 5b (blue), 6a (green), and 6b (red).
Figure 5: Chiroptical properties of oxaza[n]helicenes: (A) CD spectra measured in chloroform (1 × 10−5 M); CP...
Subsequently, CPL spectra of (P/M)-5a,b and (P/M)-6a,b were measured to evaluate the potential of these oxaza[n]helicenes as chiral emitters. The |glum| values were determined to be 0.001 at 498.0 nm for 5a, 0.0026 at 474.5 nm for 5b, 0.0006 at 441.8 nm for 6a, and 0.0018 at 448.8 nm for 6b, with the (P)-configuration exhibiting a positive Cotton effect and the (M)-configuration showing a negative Cotton effect (Figure 5B). According to the theory [66], the luminescence dissymmetry factor |glum| can be determined by Equation 1:
![[1860-5397-22-25-i3]](/bjoc/content/inline/1860-5397-22-25-i3.svg?max-width=590&scale=1.18182)
Therefore, the electric transition dipole moments (ETDM) (μ) and magnetic transition dipole moments (MTDM) (m), as well as the angle (θ) between μ and m, of (M)-5a,b and (M)-6a,b for their S1 → S0 transitions were obtained by TD-DFT calculations (see Supporting Information File 1). For most organic CPL-emitters, the |m| values are typically much smaller than the |μ| values (Table 1). The above equation can thus be simplified as Equation 2:
![[1860-5397-22-25-i4]](/bjoc/content/inline/1860-5397-22-25-i4.svg?max-width=590&scale=1.18182)
Hence, the lower |μ| and larger cos θ values of 5b lead to an approximately 1.5-fold increase in their calculated gcal compared to corresponding oxaza[7]helicene 6b and around 2.5-fold increase in gcal compared to the unsubstituted oxaza[8]helicene 5a (Table 1), which is consistent with the trend observed experimentally (Figure 5B). With the chiroptical results and Φf in hand, the brightness BCPL values were calculated to be 30.75 M−1 cm−1 for 5b and 31.46 M−1 cm−1 for 6b. This comprehensive understanding of the influence of phenyl substitution and helical extension on the CPL features of oxaza[n]helicenes provides a valuable roadmap for designing future CPL- emitters that integrate synthetic accessibility with superior chiral stability and chiroptical performance.
Table 1: Chiroptical features of oxaza[8]helicenes 5 and oxaza[7]helicenes 6.
| S0 → S1 transition | CPL | |||||||
| Oxaza[n]helicene | ETDMa |μ| (10−20 esu cm) | MTDMb |m| (10−20 erg G−1) | θμ,m (deg)c | (R)d (10−40 erg esu cm G−1) | gcale (10−3) | λem (nm) | glume (10−3)f |
BCPL
M−1 cm−1 |
| (M)-5a | 553.8 | 1.20 | 95.9 | −68.71 | −0.90 | 498.0 | −1.0 | 15.98 |
| (M)-5b | 502.4 | 1.81 | 98.7 | −137.30 | −2.18 | 474.5 | −2.6 | 30.75 |
| (M)-6a | 665.4 | 1.06 | 94.5 | −54.84 | −0.50 | 441.8 | −0.6 | 6.13 |
| (M)-6b | 569.0 | 1.87 | 96.5 | −121.02 | −1.50 | 448.8 | −1.8 | 31.46 |
aElectric transition dipole moments (ETDM) for the S1 → S0 transitions; bmagnetic transition dipole moments (MTDM) for the S1 → S0 transitions; cthe angle between ETDM and MTDM vectors; drotational strength; edimensionless values calculated at the MN15/lanl2mb (iefpcm = chloroform); fmeasured dissymmetry factors.
Conclusion
In summary, we have established an electrosynthetic strategy to access a novel class of unsymmetrical oxaza[8]helicenes by exploiting the differential oxidation potentials of appropriately designed coupling partners to control both chemo- and regioselectivity. A finely tuned anodic sequence enables selective oxidative hetero-coupling followed by dehydrative cyclization, delivering extended [8]helical scaffolds efficiently under mild, oxidant-free conditions. Combined experimental and DFT analyses reveal that these oxaza[8]helicenes retain the aromatic character of the π‐framework while exhibiting significantly enhanced configurational stability compared to their oxaza[7] congeners, with enantiomerization barriers of up to ≈38 kcal mol−1. After chiral separation, the enantiomers display intense chiroptical responses, including mirror-image CD and strong CPL with |glum| values up to 0.0026 and CPL brightness approaching 30.8 M−1 cm−1.
Supporting Information
| Supporting Information File 1: Experimental procedures, synthetic details, NMR spectra, chiral HPLC chromatograms, DFT and TD-DFT calculations. | ||
| Format: PDF | Size: 6.8 MB | Download |
| Supporting Information File 2: Cartesian coordinates of DFT calculations. | ||
| Format: XLSX | Size: 2.1 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Peluso, P.; Chankvetadze, B. Chem. Rev. 2022, 122, 13235–13400. doi:10.1021/acs.chemrev.1c00846
Return to citation in text: [1] -
Frédéric, L.; Desmarchelier, A.; Favereau, L.; Pieters, G. Adv. Funct. Mater. 2021, 31, 2010281. doi:10.1002/adfm.202010281
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2013, 42, 1051–1095. doi:10.1039/c2cs35134j
Return to citation in text: [1] -
Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r
Return to citation in text: [1] -
Gingras, M.; Félix, G.; Peresutti, R. Chem. Soc. Rev. 2013, 42, 1007–1050. doi:10.1039/c2cs35111k
Return to citation in text: [1] -
Matsuo, Y.; Gon, M.; Tanaka, K.; Seki, S.; Tanaka, T. J. Am. Chem. Soc. 2024, 146, 17428–17437. doi:10.1021/jacs.4c05156
Return to citation in text: [1] -
Zhu, K.-L.; Li, Z.-A.; Liang, J.; Zou, K.-L.; Shen, Y.-J.; Gong, H.-Y. Angew. Chem., Int. Ed. 2024, 63, e202409713. doi:10.1002/anie.202409713
Return to citation in text: [1] [2] [3] -
Shen, Y.-J.; Yao, N.-T.; Diao, L.-N.; Yang, Y.; Chen, X.-L.; Gong, H.-Y. Angew. Chem., Int. Ed. 2023, 62, e202300840. doi:10.1002/anie.202300840
Return to citation in text: [1] -
Zhao, W.-L.; Li, M.; Lu, H.-Y.; Chen, C.-F. Chem. Commun. 2019, 55, 13793–13803. doi:10.1039/c9cc06861a
Return to citation in text: [1] -
Zhang, L.; Song, I.; Ahn, J.; Han, M.; Linares, M.; Surin, M.; Zhang, H.-J.; Oh, J. H.; Lin, J. Nat. Commun. 2021, 12, 142. doi:10.1038/s41467-020-20390-y
Return to citation in text: [1] -
Ma, Z.; Winands, T.; Liang, N.; Meng, D.; Jiang, W.; Doltsinis, N. L.; Wang, Z. Sci. China: Chem. 2020, 63, 208–214. doi:10.1007/s11426-019-9632-2
Return to citation in text: [1] -
Kelly, T. R. Acc. Chem. Res. 2001, 34, 514–522. doi:10.1021/ar000167x
Return to citation in text: [1] -
Yamamoto, K.; Ikeda, T.; Kitsuki, T.; Okamoto, Y.; Chikamatsu, H.; Nakazaki, M. J. Chem. Soc., Perkin Trans. 1 1990, 271–276. doi:10.1039/p19900000271
Return to citation in text: [1] -
Wu, Y.-F.; Ying, S.-W.; Su, L.-Y.; Du, J.-J.; Zhang, L.; Chen, B.-W.; Tian, H.-R.; Xu, H.; Zhang, M.-L.; Yan, X.; Zhang, Q.; Xie, S.-Y.; Zheng, L.-S. J. Am. Chem. Soc. 2022, 144, 10736–10742. doi:10.1021/jacs.2c00794
Return to citation in text: [1] -
Sapir, M.; Vander Donckt, E. Chem. Phys. Lett. 1975, 36, 108–110. doi:10.1016/0009-2614(75)85698-3
Return to citation in text: [1] -
Salem, M. S. H.; Takizawa, S. Chem.: Methods 2025, 5, e202500018. doi:10.1002/cmtd.202500018
Return to citation in text: [1] -
Gan, Z.; Lai, J.; Lai, L.; Xie, S.-Y.; Zhang, Q. Adv. Opt. Mater. 2025, 13, e00976. doi:10.1002/adom.202500976
Return to citation in text: [1] -
Qiu, M.; Du, J.; Yao, N.-T.; Wang, X.-Y.; Gong, H.-Y. Beilstein J. Org. Chem. 2025, 21, 1422–1453. doi:10.3762/bjoc.21.106
Return to citation in text: [1] -
Wang, X.-Y.; Yao, X.; Narita, A.; Müllen, K. Acc. Chem. Res. 2019, 52, 2491–2505. doi:10.1021/acs.accounts.9b00322
Return to citation in text: [1] -
Tan, D.; Dong, J.; Ma, T.; Feng, Q.; Wang, S.; Yang, D.-T. Angew. Chem., Int. Ed. 2023, 62, e202304711. doi:10.1002/anie.202304711
Return to citation in text: [1] -
Dhbaibi, K.; Favereau, L.; Crassous, J. Chem. Rev. 2019, 119, 8846–8953. doi:10.1021/acs.chemrev.9b00033
Return to citation in text: [1] -
Maeda, C.; Ema, T. Chem. Commun. 2025, 61, 4757–4773. doi:10.1039/d4cc06307d
Return to citation in text: [1] -
Ryan, S. T.; Fuchter, M. J. Helicenes for optoelectronic applications and devices. In Helicenes: Synthesis, Properties and Applications; Crassous, J.; Stará, I. G.; Stary, I., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp 473–503. doi:10.1002/9783527829415.ch15
Return to citation in text: [1] -
Ye, Z.; Wu, H.; Xu, Y.; Hua, T.; Chen, G.; Chen, Z.; Yin, X.; Huang, M.; Xu, K.; Song, X.; Huang, Z.; Lv, X.; Miao, J.; Cao, X.; Yang, C. Adv. Mater. (Weinheim, Ger.) 2024, 36, 2308314. doi:10.1002/adma.202308314
Return to citation in text: [1] -
Nowak-Król, A.; Geppert, P. T.; Naveen, K. R. Chem. Sci. 2024, 15, 7408–7440. doi:10.1039/d4sc01083c
Return to citation in text: [1] -
Khalid, M. I.; Salem, M. S. H.; Takizawa, S. Molecules 2024, 29, 296. doi:10.3390/molecules29020296
Return to citation in text: [1] -
Salem, M. S. H.; Sharma, R.; Suzuki, S.; Imai, Y.; Arisawa, M.; Takizawa, S. Chirality 2024, 36, e23673. doi:10.1002/chir.23673
Return to citation in text: [1] [2] [3] -
Qiu, Z.; Ju, C.-W.; Frédéric, L.; Hu, Y.; Schollmeyer, D.; Pieters, G.; Müllen, K.; Narita, A. J. Am. Chem. Soc. 2021, 143, 4661–4667. doi:10.1021/jacs.0c13197
Return to citation in text: [1] -
Full, F.; Wölflick, Q.; Radacki, K.; Braunschweig, H.; Nowak‐Król, A. Chem. – Eur. J. 2022, 28, e202202280. doi:10.1002/chem.202202280
Return to citation in text: [1] -
Fox, J. M.; Katz, T. J. J. Org. Chem. 1999, 64, 302–305. doi:10.1021/jo9817570
Return to citation in text: [1] -
Nakai, Y.; Mori, T.; Inoue, Y. J. Phys. Chem. A 2012, 116, 7372–7385. doi:10.1021/jp304576g
Return to citation in text: [1] -
Yanagi, T.; Tanaka, T.; Yorimitsu, H. Chem. Sci. 2021, 12, 2784–2793. doi:10.1039/d1sc00044f
Return to citation in text: [1] -
Yanagi, T.; Otsuka, S.; Kasuga, Y.; Fujimoto, K.; Murakami, K.; Nogi, K.; Yorimitsu, H.; Osuka, A. J. Am. Chem. Soc. 2016, 138, 14582–14585. doi:10.1021/jacs.6b10278
Return to citation in text: [1] -
Khot, S.; Khose, V.; Gavali, A.; Hasan, M.; Badani, P.; Karnik, A. J. Org. Chem. 2024, 89, 15834–15841. doi:10.1021/acs.joc.4c01971
Return to citation in text: [1] -
Yu, Y.; Wang, C.; Hung, F.-F.; Chen, C.; Pan, D.; Che, C.-M.; Liu, J. J. Am. Chem. Soc. 2024, 146, 22600–22611. doi:10.1021/jacs.4c06997
Return to citation in text: [1] -
Takizawa, S.; Salem, M. S. H., Eds. Atropisomerism in Asymmetric Organic Synthesis: Challenges and Applications; Wiley-VCH: Weinheim, Germany, 2025. doi:10.1002/9783527844258
Return to citation in text: [1] -
Fan, D.; Karuppasamy, M.; Kamble, G. T.; Ando, K.; Zhou, D.; Salem, M. S. H.; Sasai, H.; Takizawa, S. ACS Catal. 2025, 15, 18077–18086. doi:10.1021/acscatal.5c05038
Return to citation in text: [1] -
Kamble, G. T.; Salem, M. S. H.; Abe, T.; Park, H.; Sako, M.; Takizawa, S.; Sasai, H. Chem. Lett. 2021, 50, 1755–1757. doi:10.1246/cl.210367
Return to citation in text: [1] -
Li, C.; Zhou, J.; Dai, H.; Li, M.; Zhang, D.; Duan, L. InfoMat 2025, 7, e12652. doi:10.1002/inf2.12652
Return to citation in text: [1] [2] -
Yavari, K.; Retailleau, P.; Voituriez, A.; Marinetti, A. Chem. – Eur. J. 2013, 19, 9939–9947. doi:10.1002/chem.201300844
Return to citation in text: [1] [2] -
Shen, C.; Srebro‐Hooper, M.; Jean, M.; Vanthuyne, N.; Toupet, L.; Williams, J. A. G.; Torres, A. R.; Riives, A. J.; Muller, G.; Autschbach, J.; Crassous, J. Chem. – Eur. J. 2017, 23, 407–418. doi:10.1002/chem.201604398
Return to citation in text: [1] [2] -
Huang, T.; Yuan, L.; Lu, X.; Qu, Y.; Qu, C.; Xu, Y.; Zheng, Y.-X.; Wang, Y. Chem. Sci. 2024, 15, 15170–15177. doi:10.1039/d4sc03854a
Return to citation in text: [1] [2] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Zhu, C.; Ang, N. W. J.; Meyer, T. H.; Qiu, Y.; Ackermann, L. ACS Cent. Sci. 2021, 7, 415–431. doi:10.1021/acscentsci.0c01532
Return to citation in text: [1] -
Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f
Return to citation in text: [1] -
Röckl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45–61. doi:10.1021/acs.accounts.9b00511
Return to citation in text: [1] [2] -
Chen, G.; Li, X.; Feng, X. Angew. Chem., Int. Ed. 2022, 61, e202209014. doi:10.1002/anie.202209014
Return to citation in text: [1] -
Gabr, A. S.; Salem, M. S. H.; Khalid, M. I.; Takahashi, R.; Nishimoto, Y.; Yasuda, M.; Takizawa, S. Nat. Commun. 2025, 16, 5682. doi:10.1038/s41467-025-60889-w
Return to citation in text: [1] [2] -
Salem, M. S. H.; Khalid, M. I.; Sako, M.; Higashida, K.; Lacroix, C.; Kondo, M.; Takishima, R.; Taniguchi, T.; Miura, M.; Vo‐Thanh, G.; Sasai, H.; Takizawa, S. Adv. Synth. Catal. 2023, 365, 373–380. doi:10.1002/adsc.202201262
Return to citation in text: [1] [2] [3] -
Khalid, M. I.; Salem, M. S. H.; Sako, M.; Kondo, M.; Sasai, H.; Takizawa, S. Commun. Chem. 2022, 5, 166. doi:10.1038/s42004-022-00780-7
Return to citation in text: [1] -
Salem, M. S. H.; Sharma, R.; Khalid, M. I.; Sasi, M.; Amasaki, R.; Imai, Y.; Arisawa, M.; Takizawa, S. Electrochemistry 2023, 91, 112015. doi:10.5796/electrochemistry.23-67092
Return to citation in text: [1] -
Salem, M. S. H.; Sabri, A.; Khalid, M. I.; Sasai, H.; Takizawa, S. Molecules 2022, 27, 9068. doi:10.3390/molecules27249068
Return to citation in text: [1] -
Pushkarskaya, E.; Wong, B.; Han, C.; Capomolla, S.; Gu, C.; Stoltz, B. M.; Zhang, H. Tetrahedron Lett. 2016, 57, 5653–5657. doi:10.1016/j.tetlet.2016.11.009
Return to citation in text: [1] -
Salem, M. S. H.; Khalid, M. I.; Sasai, H.; Takizawa, S. Tetrahedron 2023, 133, 133266. doi:10.1016/j.tet.2023.133266
Return to citation in text: [1] -
Dorel, R.; Grugel, C. P.; Haydl, A. M. Angew. Chem., Int. Ed. 2019, 58, 17118–17129. doi:10.1002/anie.201904795
Return to citation in text: [1] -
Salem, M. S. H.; El Samak, M.; Abdel Aziz, Y. M.; Aboutaleb, M. H.; Patil, S.; Aye, T. Z.; Ibrahim, T. S.; Takizawa, S. ACS Omega 2025, 10, 59183–59199. doi:10.1021/acsomega.5c08465
Return to citation in text: [1] -
Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer, P. v. R. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+
Return to citation in text: [1] -
Schleyer, P. v. R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N. J. R. J. Am. Chem. Soc. 1996, 118, 6317–6318. doi:10.1021/ja960582d
Return to citation in text: [1] -
Johnson, C. E., Jr.; Bovey, F. A. J. Chem. Phys. 1958, 29, 1012–1014. doi:10.1063/1.1744645
Return to citation in text: [1] -
Krygowski, T. M.; Cyrañski, M. K.; Czarnocki, Z.; Häfelinger, G.; Katritzky, A. R. Tetrahedron 2000, 56, 1783–1796. doi:10.1016/s0040-4020(99)00979-5
Return to citation in text: [1] -
Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Chem. Rev. 2005, 105, 3758–3772. doi:10.1021/cr0300901
Return to citation in text: [1] -
Kubo, H.; Shimizu, D.; Hirose, T.; Matsuda, K. Org. Lett. 2020, 22, 9276–9281. doi:10.1021/acs.orglett.0c03506
Return to citation in text: [1] -
Kubo, H.; Hirose, T.; Nakashima, T.; Kawai, T.; Hasegawa, J.-y.; Matsuda, K. J. Phys. Chem. Lett. 2021, 12, 686–695. doi:10.1021/acs.jpclett.0c03174
Return to citation in text: [1] -
Hirose, T.; Tsunoi, Y.; Fujimori, Y.; Matsuda, K. Chem. – Eur. J. 2015, 21, 1637–1644. doi:10.1002/chem.201404745
Return to citation in text: [1] -
Qiu, S.; Valdivia, A. C.; Zhuang, W.; Hung, F.-F.; Che, C.-M.; Casado, J.; Liu, J. J. Am. Chem. Soc. 2024, 146, 16161–16172. doi:10.1021/jacs.4c03815
Return to citation in text: [1] -
Schellman, J. A. Chem. Rev. 1975, 75, 323–331. doi:10.1021/cr60295a004
Return to citation in text: [1]
| 48. | Gabr, A. S.; Salem, M. S. H.; Khalid, M. I.; Takahashi, R.; Nishimoto, Y.; Yasuda, M.; Takizawa, S. Nat. Commun. 2025, 16, 5682. doi:10.1038/s41467-025-60889-w |
| 56. | Salem, M. S. H.; El Samak, M.; Abdel Aziz, Y. M.; Aboutaleb, M. H.; Patil, S.; Aye, T. Z.; Ibrahim, T. S.; Takizawa, S. ACS Omega 2025, 10, 59183–59199. doi:10.1021/acsomega.5c08465 |
| 46. | Röckl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45–61. doi:10.1021/acs.accounts.9b00511 |
| 57. | Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer, P. v. R. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+ |
| 58. | Schleyer, P. v. R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N. J. R. J. Am. Chem. Soc. 1996, 118, 6317–6318. doi:10.1021/ja960582d |
| 1. | Peluso, P.; Chankvetadze, B. Chem. Rev. 2022, 122, 13235–13400. doi:10.1021/acs.chemrev.1c00846 |
| 2. | Frédéric, L.; Desmarchelier, A.; Favereau, L.; Pieters, G. Adv. Funct. Mater. 2021, 31, 2010281. doi:10.1002/adfm.202010281 |
| 34. | Khot, S.; Khose, V.; Gavali, A.; Hasan, M.; Badani, P.; Karnik, A. J. Org. Chem. 2024, 89, 15834–15841. doi:10.1021/acs.joc.4c01971 |
| 62. | Kubo, H.; Shimizu, D.; Hirose, T.; Matsuda, K. Org. Lett. 2020, 22, 9276–9281. doi:10.1021/acs.orglett.0c03506 |
| 63. | Kubo, H.; Hirose, T.; Nakashima, T.; Kawai, T.; Hasegawa, J.-y.; Matsuda, K. J. Phys. Chem. Lett. 2021, 12, 686–695. doi:10.1021/acs.jpclett.0c03174 |
| 10. | Zhang, L.; Song, I.; Ahn, J.; Han, M.; Linares, M.; Surin, M.; Zhang, H.-J.; Oh, J. H.; Lin, J. Nat. Commun. 2021, 12, 142. doi:10.1038/s41467-020-20390-y |
| 11. | Ma, Z.; Winands, T.; Liang, N.; Meng, D.; Jiang, W.; Doltsinis, N. L.; Wang, Z. Sci. China: Chem. 2020, 63, 208–214. doi:10.1007/s11426-019-9632-2 |
| 35. | Yu, Y.; Wang, C.; Hung, F.-F.; Chen, C.; Pan, D.; Che, C.-M.; Liu, J. J. Am. Chem. Soc. 2024, 146, 22600–22611. doi:10.1021/jacs.4c06997 |
| 64. | Hirose, T.; Tsunoi, Y.; Fujimori, Y.; Matsuda, K. Chem. – Eur. J. 2015, 21, 1637–1644. doi:10.1002/chem.201404745 |
| 6. | Matsuo, Y.; Gon, M.; Tanaka, K.; Seki, S.; Tanaka, T. J. Am. Chem. Soc. 2024, 146, 17428–17437. doi:10.1021/jacs.4c05156 |
| 7. | Zhu, K.-L.; Li, Z.-A.; Liang, J.; Zou, K.-L.; Shen, Y.-J.; Gong, H.-Y. Angew. Chem., Int. Ed. 2024, 63, e202409713. doi:10.1002/anie.202409713 |
| 8. | Shen, Y.-J.; Yao, N.-T.; Diao, L.-N.; Yang, Y.; Chen, X.-L.; Gong, H.-Y. Angew. Chem., Int. Ed. 2023, 62, e202300840. doi:10.1002/anie.202300840 |
| 9. | Zhao, W.-L.; Li, M.; Lu, H.-Y.; Chen, C.-F. Chem. Commun. 2019, 55, 13793–13803. doi:10.1039/c9cc06861a |
| 31. | Nakai, Y.; Mori, T.; Inoue, Y. J. Phys. Chem. A 2012, 116, 7372–7385. doi:10.1021/jp304576g |
| 61. | Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Chem. Rev. 2005, 105, 3758–3772. doi:10.1021/cr0300901 |
| 3. | Gingras, M. Chem. Soc. Rev. 2013, 42, 1051–1095. doi:10.1039/c2cs35134j |
| 4. | Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r |
| 5. | Gingras, M.; Félix, G.; Peresutti, R. Chem. Soc. Rev. 2013, 42, 1007–1050. doi:10.1039/c2cs35111k |
| 32. | Yanagi, T.; Tanaka, T.; Yorimitsu, H. Chem. Sci. 2021, 12, 2784–2793. doi:10.1039/d1sc00044f |
| 33. | Yanagi, T.; Otsuka, S.; Kasuga, Y.; Fujimoto, K.; Murakami, K.; Nogi, K.; Yorimitsu, H.; Osuka, A. J. Am. Chem. Soc. 2016, 138, 14582–14585. doi:10.1021/jacs.6b10278 |
| 27. | Salem, M. S. H.; Sharma, R.; Suzuki, S.; Imai, Y.; Arisawa, M.; Takizawa, S. Chirality 2024, 36, e23673. doi:10.1002/chir.23673 |
| 16. | Salem, M. S. H.; Takizawa, S. Chem.: Methods 2025, 5, e202500018. doi:10.1002/cmtd.202500018 |
| 17. | Gan, Z.; Lai, J.; Lai, L.; Xie, S.-Y.; Zhang, Q. Adv. Opt. Mater. 2025, 13, e00976. doi:10.1002/adom.202500976 |
| 18. | Qiu, M.; Du, J.; Yao, N.-T.; Wang, X.-Y.; Gong, H.-Y. Beilstein J. Org. Chem. 2025, 21, 1422–1453. doi:10.3762/bjoc.21.106 |
| 19. | Wang, X.-Y.; Yao, X.; Narita, A.; Müllen, K. Acc. Chem. Res. 2019, 52, 2491–2505. doi:10.1021/acs.accounts.9b00322 |
| 20. | Tan, D.; Dong, J.; Ma, T.; Feng, Q.; Wang, S.; Yang, D.-T. Angew. Chem., Int. Ed. 2023, 62, e202304711. doi:10.1002/anie.202304711 |
| 21. | Dhbaibi, K.; Favereau, L.; Crassous, J. Chem. Rev. 2019, 119, 8846–8953. doi:10.1021/acs.chemrev.9b00033 |
| 22. | Maeda, C.; Ema, T. Chem. Commun. 2025, 61, 4757–4773. doi:10.1039/d4cc06307d |
| 27. | Salem, M. S. H.; Sharma, R.; Suzuki, S.; Imai, Y.; Arisawa, M.; Takizawa, S. Chirality 2024, 36, e23673. doi:10.1002/chir.23673 |
| 28. | Qiu, Z.; Ju, C.-W.; Frédéric, L.; Hu, Y.; Schollmeyer, D.; Pieters, G.; Müllen, K.; Narita, A. J. Am. Chem. Soc. 2021, 143, 4661–4667. doi:10.1021/jacs.0c13197 |
| 29. | Full, F.; Wölflick, Q.; Radacki, K.; Braunschweig, H.; Nowak‐Król, A. Chem. – Eur. J. 2022, 28, e202202280. doi:10.1002/chem.202202280 |
| 59. | Johnson, C. E., Jr.; Bovey, F. A. J. Chem. Phys. 1958, 29, 1012–1014. doi:10.1063/1.1744645 |
| 15. | Sapir, M.; Vander Donckt, E. Chem. Phys. Lett. 1975, 36, 108–110. doi:10.1016/0009-2614(75)85698-3 |
| 30. | Fox, J. M.; Katz, T. J. J. Org. Chem. 1999, 64, 302–305. doi:10.1021/jo9817570 |
| 60. | Krygowski, T. M.; Cyrañski, M. K.; Czarnocki, Z.; Häfelinger, G.; Katritzky, A. R. Tetrahedron 2000, 56, 1783–1796. doi:10.1016/s0040-4020(99)00979-5 |
| 14. | Wu, Y.-F.; Ying, S.-W.; Su, L.-Y.; Du, J.-J.; Zhang, L.; Chen, B.-W.; Tian, H.-R.; Xu, H.; Zhang, M.-L.; Yan, X.; Zhang, Q.; Xie, S.-Y.; Zheng, L.-S. J. Am. Chem. Soc. 2022, 144, 10736–10742. doi:10.1021/jacs.2c00794 |
| 7. | Zhu, K.-L.; Li, Z.-A.; Liang, J.; Zou, K.-L.; Shen, Y.-J.; Gong, H.-Y. Angew. Chem., Int. Ed. 2024, 63, e202409713. doi:10.1002/anie.202409713 |
| 13. | Yamamoto, K.; Ikeda, T.; Kitsuki, T.; Okamoto, Y.; Chikamatsu, H.; Nakazaki, M. J. Chem. Soc., Perkin Trans. 1 1990, 271–276. doi:10.1039/p19900000271 |
| 23. | Ryan, S. T.; Fuchter, M. J. Helicenes for optoelectronic applications and devices. In Helicenes: Synthesis, Properties and Applications; Crassous, J.; Stará, I. G.; Stary, I., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp 473–503. doi:10.1002/9783527829415.ch15 |
| 24. | Ye, Z.; Wu, H.; Xu, Y.; Hua, T.; Chen, G.; Chen, Z.; Yin, X.; Huang, M.; Xu, K.; Song, X.; Huang, Z.; Lv, X.; Miao, J.; Cao, X.; Yang, C. Adv. Mater. (Weinheim, Ger.) 2024, 36, 2308314. doi:10.1002/adma.202308314 |
| 25. | Nowak-Król, A.; Geppert, P. T.; Naveen, K. R. Chem. Sci. 2024, 15, 7408–7440. doi:10.1039/d4sc01083c |
| 26. | Khalid, M. I.; Salem, M. S. H.; Takizawa, S. Molecules 2024, 29, 296. doi:10.3390/molecules29020296 |
| 49. | Salem, M. S. H.; Khalid, M. I.; Sako, M.; Higashida, K.; Lacroix, C.; Kondo, M.; Takishima, R.; Taniguchi, T.; Miura, M.; Vo‐Thanh, G.; Sasai, H.; Takizawa, S. Adv. Synth. Catal. 2023, 365, 373–380. doi:10.1002/adsc.202201262 |
| 39. | Li, C.; Zhou, J.; Dai, H.; Li, M.; Zhang, D.; Duan, L. InfoMat 2025, 7, e12652. doi:10.1002/inf2.12652 |
| 36. | Takizawa, S.; Salem, M. S. H., Eds. Atropisomerism in Asymmetric Organic Synthesis: Challenges and Applications; Wiley-VCH: Weinheim, Germany, 2025. doi:10.1002/9783527844258 |
| 37. | Fan, D.; Karuppasamy, M.; Kamble, G. T.; Ando, K.; Zhou, D.; Salem, M. S. H.; Sasai, H.; Takizawa, S. ACS Catal. 2025, 15, 18077–18086. doi:10.1021/acscatal.5c05038 |
| 38. | Kamble, G. T.; Salem, M. S. H.; Abe, T.; Park, H.; Sako, M.; Takizawa, S.; Sasai, H. Chem. Lett. 2021, 50, 1755–1757. doi:10.1246/cl.210367 |
| 49. | Salem, M. S. H.; Khalid, M. I.; Sako, M.; Higashida, K.; Lacroix, C.; Kondo, M.; Takishima, R.; Taniguchi, T.; Miura, M.; Vo‐Thanh, G.; Sasai, H.; Takizawa, S. Adv. Synth. Catal. 2023, 365, 373–380. doi:10.1002/adsc.202201262 |
| 39. | Li, C.; Zhou, J.; Dai, H.; Li, M.; Zhang, D.; Duan, L. InfoMat 2025, 7, e12652. doi:10.1002/inf2.12652 |
| 40. | Yavari, K.; Retailleau, P.; Voituriez, A.; Marinetti, A. Chem. – Eur. J. 2013, 19, 9939–9947. doi:10.1002/chem.201300844 |
| 41. | Shen, C.; Srebro‐Hooper, M.; Jean, M.; Vanthuyne, N.; Toupet, L.; Williams, J. A. G.; Torres, A. R.; Riives, A. J.; Muller, G.; Autschbach, J.; Crassous, J. Chem. – Eur. J. 2017, 23, 407–418. doi:10.1002/chem.201604398 |
| 42. | Huang, T.; Yuan, L.; Lu, X.; Qu, Y.; Qu, C.; Xu, Y.; Zheng, Y.-X.; Wang, Y. Chem. Sci. 2024, 15, 15170–15177. doi:10.1039/d4sc03854a |
| 27. | Salem, M. S. H.; Sharma, R.; Suzuki, S.; Imai, Y.; Arisawa, M.; Takizawa, S. Chirality 2024, 36, e23673. doi:10.1002/chir.23673 |
| 65. | Qiu, S.; Valdivia, A. C.; Zhuang, W.; Hung, F.-F.; Che, C.-M.; Casado, J.; Liu, J. J. Am. Chem. Soc. 2024, 146, 16161–16172. doi:10.1021/jacs.4c03815 |
| 54. | Salem, M. S. H.; Khalid, M. I.; Sasai, H.; Takizawa, S. Tetrahedron 2023, 133, 133266. doi:10.1016/j.tet.2023.133266 |
| 55. | Dorel, R.; Grugel, C. P.; Haydl, A. M. Angew. Chem., Int. Ed. 2019, 58, 17118–17129. doi:10.1002/anie.201904795 |
| 48. | Gabr, A. S.; Salem, M. S. H.; Khalid, M. I.; Takahashi, R.; Nishimoto, Y.; Yasuda, M.; Takizawa, S. Nat. Commun. 2025, 16, 5682. doi:10.1038/s41467-025-60889-w |
| 49. | Salem, M. S. H.; Khalid, M. I.; Sako, M.; Higashida, K.; Lacroix, C.; Kondo, M.; Takishima, R.; Taniguchi, T.; Miura, M.; Vo‐Thanh, G.; Sasai, H.; Takizawa, S. Adv. Synth. Catal. 2023, 365, 373–380. doi:10.1002/adsc.202201262 |
| 50. | Khalid, M. I.; Salem, M. S. H.; Sako, M.; Kondo, M.; Sasai, H.; Takizawa, S. Commun. Chem. 2022, 5, 166. doi:10.1038/s42004-022-00780-7 |
| 51. | Salem, M. S. H.; Sharma, R.; Khalid, M. I.; Sasi, M.; Amasaki, R.; Imai, Y.; Arisawa, M.; Takizawa, S. Electrochemistry 2023, 91, 112015. doi:10.5796/electrochemistry.23-67092 |
| 52. | Salem, M. S. H.; Sabri, A.; Khalid, M. I.; Sasai, H.; Takizawa, S. Molecules 2022, 27, 9068. doi:10.3390/molecules27249068 |
| 53. | Pushkarskaya, E.; Wong, B.; Han, C.; Capomolla, S.; Gu, C.; Stoltz, B. M.; Zhang, H. Tetrahedron Lett. 2016, 57, 5653–5657. doi:10.1016/j.tetlet.2016.11.009 |
| 42. | Huang, T.; Yuan, L.; Lu, X.; Qu, Y.; Qu, C.; Xu, Y.; Zheng, Y.-X.; Wang, Y. Chem. Sci. 2024, 15, 15170–15177. doi:10.1039/d4sc03854a |
| 43. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 44. | Zhu, C.; Ang, N. W. J.; Meyer, T. H.; Qiu, Y.; Ackermann, L. ACS Cent. Sci. 2021, 7, 415–431. doi:10.1021/acscentsci.0c01532 |
| 45. | Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Chem. Soc. Rev. 2021, 50, 7941–8002. doi:10.1039/d1cs00223f |
| 46. | Röckl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45–61. doi:10.1021/acs.accounts.9b00511 |
| 47. | Chen, G.; Li, X.; Feng, X. Angew. Chem., Int. Ed. 2022, 61, e202209014. doi:10.1002/anie.202209014 |
| 40. | Yavari, K.; Retailleau, P.; Voituriez, A.; Marinetti, A. Chem. – Eur. J. 2013, 19, 9939–9947. doi:10.1002/chem.201300844 |
| 41. | Shen, C.; Srebro‐Hooper, M.; Jean, M.; Vanthuyne, N.; Toupet, L.; Williams, J. A. G.; Torres, A. R.; Riives, A. J.; Muller, G.; Autschbach, J.; Crassous, J. Chem. – Eur. J. 2017, 23, 407–418. doi:10.1002/chem.201604398 |
| 7. | Zhu, K.-L.; Li, Z.-A.; Liang, J.; Zou, K.-L.; Shen, Y.-J.; Gong, H.-Y. Angew. Chem., Int. Ed. 2024, 63, e202409713. doi:10.1002/anie.202409713 |
© 2026 Aye et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.