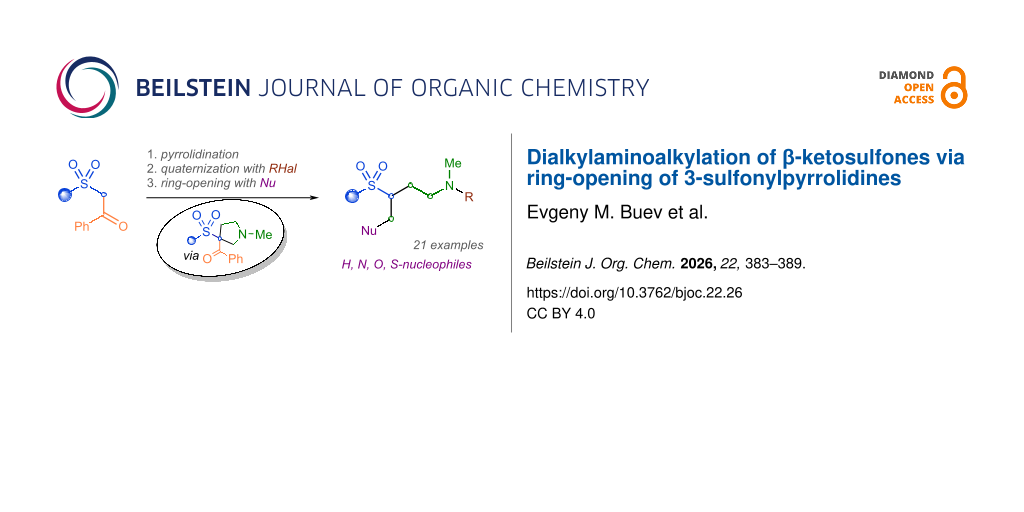
Abstract
Herein, a three-step method for the simultaneous dialkylaminoethylation and heteromethylation of active methylene β-ketosulfones, promoted by a leaving benzoyl group, is proposed. The primary domino reaction of sarcosine, paraformaldehyde, and β-ketosulfones affords 3-sulfonyl-3-benzoylpyrrolidines in good to high yields. Further treatment with alkyl halides and heating of the quaternary ammonium salt in the presence of a O-, S- or N-nucleophile with cesium carbonate leads to a retro-Claisen reaction, ring-opening and Michael addition. The resulting novel 4-Nu-3-sulfonylbutan-1-amines are isolated in moderate to excellent overall yields. The reduction of the obtained products with LiAlH4 leads to a substitution of the attached nucleophilic moiety with hydrogen in good yields.

Graphical Abstract
Introduction
Sulfones and sulfonic acids are valuable structural motifs in organic chemistry, notable for their broad spectrum of pharmacological activities [1-3] and synthetic potential [4,5]. Also noteworthy are γ-aminosulfones, which form the backbone of important pharmaceuticals including the antiglaucoma drug dorzolamide [6], acamprosate for alcohol craving suppression [7], and bicalutamide, an antiandrogen cancer treatment [8,9] (Figure 1). Moreover, aminosulfones remain a privileged scaffold in the ongoing development of new methods for the synthesis of pharmaceuticals [10-12], with multiple research compounds showing promising bioactivities such as MMP inhibition [13], antiinflammatory effects [14], сoagulation enzyme factor (FXa) inhibition [15] and antidepressant properties [16].
![[1860-5397-22-26-1]](/bjoc/content/figures/1860-5397-22-26-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Valuable amino sulfonic acids and aminosulfones.
Figure 1: Valuable amino sulfonic acids and aminosulfones.
Considering the approaches to the synthesis of γ-aminosulfones, we focused our attention on the implementation of an aminoalkylation as a powerful and versatile tool for the synthesis of aliphatic amines [17-19]. Among the various types of aminoalkylations, aminomethylation reactions have been the most extensively studied and applied significantly to aromatic [20] and CH-acidic compounds [21], alkanes [22,23], olefines [24,25] and electron-withdrawing alkenes [26,27].
This rapidly growing research field is offering diverse methodologies that enable the introduction of either protected nitrogen atoms or aliphatic amine groups. Such diversity has greatly expanded the synthetic arsenal available for constructing aminomethylated compounds.
In contrast, methods allowing an aminoethylation process is commonly achieved by a classic nucleophilic substitution of a leaving group at the β-position relative to the N-protected nitrogen (Figure 2a). While this approach addresses most needs of targeted synthesis, developing alternative methods from inexpensive starting materials via domino-processes, allowing the fast increase of the molecular complexity, remains significant. Unlike the classical aminoethylation of nucleophilic compounds [28,29], Batey et al. recently developed a two-step approach consisted in addition of terminal ynimides I to various electrophiles with subsequent reduction of the C≡C bond (Figure 2b) [30]. At the same time, Yang et al. presented a domino radical reaction of CH2-active compounds II and N,N-dimethylanilines III promoted by di-tert-butyl peroxide (DTBP), allowing the formation of the ethylene link via a domino-process of the methylenation and subsequent aminomethyl radical addition (Figure 2c) [31].
![[1860-5397-22-26-2]](/bjoc/content/figures/1860-5397-22-26-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected approaches to aminoethylation.
Figure 2: Selected approaches to aminoethylation.
In the continuation of our work on application of pyrrolidines as sacrificial framework for the alkylaminomethylation of enones [32,33] we turned our attention to active methylene compounds IV as precursors of pyrrolidines. Preliminary experiments demonstrated that the quaternary ammonium salt of 3,3-dibenzoylpyrrolidine, derived from dibenzoylmethane, undergoes ring-opening in the presence of a nucleophile and sodium methoxide via a retro-Claisen reaction [33]. This process yields 4-(dimethylamino)-2-heteromethyl-1-phenylbutan-1-one scaffold V (EWG = COPh; Y = OR, SR, NR; LG = H or COPh). We envisioned that such an approach is useful for the simultaneous introduction of aminoethyl and heteromethyl moieties at other active methylene compounds (Figure 2d). In this work, we focused on such dual aminoalkylative difunctionalization of β-ketosulfones [4,34] through a three-step method for the synthesis of N,N-dialkyl-3-(sulfonyl)butan-1-amines.
Results and Discussion
We began our survey with the examination of the pyrrolidination reaction [35,36] of β-ketosulfones (Scheme 1). This domino reaction of an active methylene compound, N-methylglycine and paraformaldehyde consists in the simultaneous generation of two reactive intermediates: terminal alkene A and N-methylazomethine ylide B, and final [3 + 2] cycloaddition. It was found that the latter approach depends on CH-acidity of the active methylene compounds and performs well with the acidic substrates in the range of pKa 12–17 (in DMSO) [37]. Considering that the pKa of 1-phenyl-2-(phenylsulfonyl)ethan-1-one 1 is close to that interval (pKa 11.4, DMSO) we estimated its smooth incorporation in the process. Nevertheless, previously developed conditions for the pyrrolidination reaction provided insufficient results. Refluxing a mixture of ketosulfone 1, a two-fold excesses of N-methylglycine and paraformaldehyde in benzene with a Dean–Stark trap for 4 h yields incomplete conversion of the substrate (Scheme 1, product 2a, 60%). However, we did not observe the resinification of the reaction media and hence we decided to increase the reaction temperature. Thus, refluxing the reagents in the PhMe/MTBE mixture (v/v 17/3, approximately 100 °C) for 4 h led to complete conversion of the ketosulfone and formation of 3-phenylsulfonyl-3-benzoyl-N-methylpyrrolidine (2a) in almost quantitative yield.
![[1860-5397-22-26-i1]](/bjoc/content/inline/1860-5397-22-26-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pyrrolidination of β-ketosulfones. Reaction conditions: β-ketosulfones (4.0 mmol), sarcosine (1.2 equiv), (CH2O)n (2.5 equiv), PhMe/MTBE (17/3 mL), reflux 4 h. a2.5 equiv of sarcosine was used.
Scheme 1: Pyrrolidination of β-ketosulfones. Reaction conditions: β-ketosulfones (4.0 mmol), sarcosine (1.2 e...
Despite that [3 + 2] cycloaddition of azomethine ylides at various vinyl sulfones were well studied [38-41], a method for the preparation of 3-sulfonylpyrrolidines from β-ketosulfones was previously unknown. We synthesized a number of pyrrolidines 2a–f in 83–97% yield, which were purified by an acid-base extraction (Scheme 1). Altering the substituents at the sulfonyl group made no significant impact on the yield of the reaction. An application of β-ethoxycarbonylsulfone lowered the yield of the pyrrolidine 2f (53%), apparently due to the lower acidity and activity of its methylene group, however, increasing the excess of azomethine ylide precursors (2.5:2.5 equiv sarcosine/CH2O) led to 87% yield.
The obtained pyrrolidines represent a class of labile compounds with a strained C2–C3 bond. The presence of the benzoyl and sulfonyl groups able to eliminate [5,42] and generate an anion at the 3rd position should promote Hofmann elimination. To enhance the leaving ability of the nitrogen atom we prepared ammonium salts 3 of the obtained pyrrolidines via treatment with alkyl halides. Since the preparation of quaternary ammonium salts of pyrrolidines is straightforward, we employed a one-pot method for its synthesis by adding the alkyl halide directly to the cooled reaction mixture of the formed pyrrolidine without its isolation and purification, that provided higher yields of the quaternary compounds 3 rather than step by step approach.
Further, we focused our attention on the search for the reaction conditions for the ring-cleavage of the obtained quaternary compound 3a. An application of sodium methylate, suitable for the 3,3-dibenzoylpyrrolidine salt [33], provided only 45% conversion of the substrate (Table 1, entry 1). Treating a methanolic solution of quaternary compound 3a with milder base such as cesium carbonate at 25 °C for 96 h provided higher yield (Table 1, entry 2, 70%). However, heating this mixture at 65 °C for 24 h in a sealed vial gave pure desired 4-methoxy-N,N-dimethyl-3-(phenylsulfonyl)butan-1-amine (4a) in 90% yield and 83% overall yield starting from ketosulfone 1a (Table 1, entry 4).
Table 1: Synthetic sequence and optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-22-26-i5.svg?max-width=637&scale=1.0)
|
||
| Ring-cleavage step conditions | Result | |
| 1 | MeONa (1.2 equiv), MeOH, 25 °C, 24 h | 4a (45%) |
| 2 | Cs2CO3 (2.1 equiv), MeOH, 25 °C, 96 h | 4a (70%) |
| 3 | Cs2CO3 (2.1 equiv), MeOH, 65 °C, 15 h | 4a (74%) |
| 4 | Cs2CO3 (2.1 equiv), MeOH, 65 °C, 24 h | 4a (90%)b |
| 5 |
CO2Et instead of COPh as a leaving group
KOH (4.2 equiv), MeOH, 65 °C, 46 h |
4a (89%)b,c |
aThe reaction sequence can be stopped at any stage to isolate the intermediate products 2 and 3. The yield of 3a is overall yield calculated on starting β-ketosulfone 1a. bIsolated yield. cOverall yield is 77%.
In attempt to minimize the size of the leaving group we applied a ethoxycarbоnyl moiety instead of the benzoyl group. Thus, heating the dimethylammonium salt of pyrrolidine 2f with an excess of KOH in MeOH for 46 h provided the same product 4a in 89% yield (Table 1, entry 5). While it is possible to use an ester group for the cleavage of 3-sulfonylpyrrolidines, it required a longer reaction time and provided a slightly lower overall product yield (77%) compared to the use of the benzoyl group.
With an optimized synthetic route in hand – comprising a multicomponent reaction of ketosulfone, sarcosine, and paraformaldehyde in PhMe/MTBE, followed by treatment of the reaction mixture with an alkyl halide, and subsequent heating of the precipitated quaternary ammonium salt in the presence of Cs₂CO₃ and a nucleophile – we proceeded to apply this methodology to other β-ketosulfones.
Next, we investigated the influence of both the substituent at the sulfonyl group and the alkyl chain introduced at the nitrogen atom. It turned out that the electron-donating groups at the aryl ring or a methyl group at the sulfonyl moiety lowered the yield and the corresponding 4-methoxy-N,N-dialkyl-3-(sulfonyl)butan-1-amines 4c–e,h were isolated in 40–69% yields (Scheme 2). Introduction of the m-nitro substituent led to partial resinification during the ring-cleavage step and significant decrease in the reaction outcome (products 4f,g). We have demonstrated that ethyl bromide, benzyl chloride, allyl bromide, and even the bulky 2-(2-bromoethyl)-1,3-dioxane, which contains a masked aldehyde moiety, are suitable alkylating agents for the process, providing products 4h–k in good overall yields. The possibility to vary alkyl halide in the second stage of the synthesis opens opportunities for further modifications and the use of the obtained compounds as building blocks.
![[1860-5397-22-26-i2]](/bjoc/content/inline/1860-5397-22-26-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the reaction. Reaction conditions: sulfone 1 (1.0 mmol), sarcosine (1.2 equiv), (CH2O)n (2.5 equiv), PhMe/MTBE (17/3 v/v), Dean–Stark trap, reflux, 4 h, cooled to rt and then AlkHal (1.5 equiv), rt, 24 h (see Supporting Information File 1 for details). Quaternary compound 3, Cs2CO3 (2.1 equiv), MeOH, 65 °C, 24 h for 4a–f,h–k,q–t or CH3CN, 80 °C, 24 h for 4g,l–p. aEthanol 95% was used instead of methanol, heating at 80 °C for 36 h. bThe quaternization with AlkHal (1.5 equiv) was performed at 70 °C for 7 days. cTHIQ = 1,2,3,4-tetrahydroisoquinolin-2-yl.
Scheme 2: Scope of the reaction. Reaction conditions: sulfone 1 (1.0 mmol), sarcosine (1.2 equiv), (CH2O)n (2...
We also examined other nucleophiles suitable for the described domino-sequence. Thus, application of ethanol as solvent resulted in a mixture of the desired product 4b with the corresponding intermediate C (NMR ratio 88:12), however, longer heating for 36 h provided ethoxy product 4b in 74% yield. Heating quaternary ammonium salt 3a with an excess of secondary amines in acetonitrile enabled concurrent aminomethylation and aminoethylation of the starting β-ketosulfone in good to excellent overall yields. Notably, even application of aqueous solution of dimethylamine did not affect the process and bis-dimethylamino product 4l was readily isolated in 97% yield. The obtained 2-(phenylsulfonyl)butane-1,4-diamines 4l–p represent the biogenic amine putrescine scaffold, incorporating dialkylamino or azaheterocyclic motifs such as pyrrolidine, piperidine, morpholine, or 1,2,3,4-tetrahydroisoquinoline (THIQ). A special curiosity was associated with the use of mercaptans as nucleophiles for the synthesis of aminobutanes 4 containing both sulfide and sulfone moieties. To our delight, an excess of butylmercaptan or thiophenol readily afforded products 4q–t in 61–87% yields.
It should be noted that the leaving 3-benzoyl group formed a methyl benzoate when the reactions were performed in methanol. We also observed the formation of S-butyl benzothioate in the reaction mixture in case of 4q. These by-products were readily removed by an acid-base extraction or column chromatography. On the other hand, upon performing the reaction with amines in acetonitrile we did not observe the formation of benzamides. Presumably, debenzoylation occurred in basic media with water traces and cesium benzoate was discarded upon work-up. The presumable mechanism of the domino-process consists in nucleophilic addition of the alcohol, mercaptan or water (in case the reaction was performed in CH3CN) at the benzoyl group (Scheme 3). The subsequent ring-opening of the pyrrolidine ring can proceed via two possible pathways. First path is the tandem Grob-type fragmentation. Second is a step-wise route through retro-Claisen reaction, formation of the carbanion at the α-position to sulfonyl group, and Hofmann elimination of the quaternary nitrogen at the β-position. Both routes lead to the formation of terminal vinyl sulfone C, which reacts with the nucleophile present in the reaction media to form 4-Nu-3-(sulfonyl)butan-1-amines 4.
![[1860-5397-22-26-i3]](/bjoc/content/inline/1860-5397-22-26-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The presumable mechanism of the domino process.
Scheme 3: The presumable mechanism of the domino process.
An attempt to obtain 3,4-bis(phenylthio)butan-1-amine via reduction of the sulfonyl group [43,44] of the product 4r with LiAlH4 gave an unexpected reductive elimination of the thiophenol moiety furnishing N,N-dimethyl-3-(phenylsulfonyl)butan-1-amine (5) in 77% yield (Scheme 4). This observation prompted us to examine methoxy and morpholine adducts 4a and 4o in the same process that resulted in the isolation of sulfonylamine 5 in 69 and 48%, respectively. The literature search revealed that previously Langler and co-authors observed the similar process of the reductive elimination of chlorine or chlorosulfonyl groups at the β-position to sulfone [45]. We also examined the starting ammonium salt 3a in the reductive ring-cleavage. As the result, butanamine 5 was formed in 18% yield along with benzylic alcohol. An increase of the reaction temperature resulted in the formation of a complex mixture with only traces of product 5. Nevertheless, the sequence of the nucleophilic ring-cleavage of the pyrrolidine ammonium salt 3 with alcohol, amine or thiol and the subsequent reduction with LiAlH4 serves as a formal reductive cleavage of the N–C2 bond of the pyrrolidines 2. In this way, the hydride anion was formally added to the list of possible nucleophiles (H-, N-, O-, S-).
![[1860-5397-22-26-i4]](/bjoc/content/inline/1860-5397-22-26-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reductive elimination of the nucleophiles.
Scheme 4: Reductive elimination of the nucleophiles.
Conclusion
In conclusion, we developed a practical route to the novel skeleton of N,N-dialkyl-3-(sulfonyl)butan-1-amines from β-ketosulfones. The proposed approach exploits pyrrolidines as sacrificial framework and two domino processes used for assembling of the pyrrolidine ring and for the ring-cleavage of it. Thus, one-pot pyrrolidination of β-ketosulfones and alkylation combined with the subsequent debenzoylative ring-cleavage allows for wide range of the incorporated reagents, that allows a broad functionalization of the 3-(sulfonyl)butan-1-amine with various substituents and their utilization as building blocks. We believe this approach has promising features for further development and pharmaceutical applications.
Supporting Information
| Supporting Information File 1: Experimental section, characterization data and copies of spectra. | ||
| Format: PDF | Size: 5.7 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Supuran, C. T.; Casini, A.; Scozzafava, A. Med. Res. Rev. 2003, 23, 535–558. doi:10.1002/med.10047
Return to citation in text: [1] -
Ahmadi, R.; Emami, S. Eur. J. Med. Chem. 2022, 234, 114255. doi:10.1016/j.ejmech.2022.114255
Return to citation in text: [1] -
Huang, H.-L.; Shi, Y.-Q.; Ning, J.-X.; Li, S.; Song, D.-T.; Gao, F.; Ma, N. Molecules 2024, 29, 5868. doi:10.3390/molecules29245868
Return to citation in text: [1] -
Trost, B. M.; Kalnmals, C. A. Chem. – Eur. J. 2019, 25, 11193–11213. doi:10.1002/chem.201902019
Return to citation in text: [1] [2] -
Nambo, M.; Maekawa, Y.; Crudden, C. M. ACS Catal. 2022, 12, 3013–3032. doi:10.1021/acscatal.1c05608
Return to citation in text: [1] [2] -
Franca, J. R.; Foureaux, G.; Fuscaldi, L. L.; Ribeiro, T. G.; Castilho, R. O.; Yoshida, M. I.; Cardoso, V. N.; Fernandes, S. O. A.; Cronemberger, S.; Nogueira, J. C.; Ferreira, A. J.; Faraco, A. A. G. Int. J. Pharm. 2019, 570, 118662. doi:10.1016/j.ijpharm.2019.118662
Return to citation in text: [1] -
Plosker, G. L. Drugs 2015, 75, 1255–1268. doi:10.1007/s40265-015-0423-9
Return to citation in text: [1] -
Tucker, H.; Crook, J. W.; Chesterson, G. J. J. Med. Chem. 1988, 31, 954–959. doi:10.1021/jm00400a011
Return to citation in text: [1] -
Parent, E. E.; Dence, C. S.; Jenks, C.; Sharp, T. L.; Welch, M. J.; Katzenellenbogen, J. A. J. Med. Chem. 2007, 50, 1028–1040. doi:10.1021/jm060847r
Return to citation in text: [1] -
Zhang, H.; Zhao, X.; Yan, R.; Lin, W. Org. Lett. 2024, 26, 4251–4256. doi:10.1021/acs.orglett.4c01103
Return to citation in text: [1] -
Qian, Z.-M.; Wang, F.-Y.; Guan, Z.; He, Y.-H. Adv. Synth. Catal. 2024, 366, 3022–3028. doi:10.1002/adsc.202400432
Return to citation in text: [1] -
Griffiths, R. J.; Kong, W. C.; Richards, S. A.; Burley, G. A.; Willis, M. C.; Talbot, E. P. A. Chem. Sci. 2018, 9, 2295–2300. doi:10.1039/c7sc04900e
Return to citation in text: [1] -
Aranapakam, V.; Davis, J. M.; Grosu, G. T.; Baker; Ellingboe, J.; Zask, A.; Levin, J. I.; Sandanayaka, V. P.; Du, M.; Skotnicki, J. S.; DiJoseph, J. F.; Sung, A.; Sharr, M. A.; Killar, L. M.; Walter, T.; Jin, G.; Cowling, R.; Tillett, J.; Zhao, W.; McDevitt, J.; Xu, Z. B. J. Med. Chem. 2003, 46, 2376–2396. doi:10.1021/jm0205550
Return to citation in text: [1] -
Al-Riyami, L.; Pineda, M. A.; Rzepecka, J.; Huggan, J. K.; Khalaf, A. I.; Suckling, C. J.; Scott, F. J.; Rodgers, D. T.; Harnett, M. M.; Harnett, W. J. Med. Chem. 2013, 56, 9982–10002. doi:10.1021/jm401251p
Return to citation in text: [1] -
Fujimoto, T.; Imaeda, Y.; Konishi, N.; Hiroe, K.; Kawamura, M.; Textor, G. P.; Aertgeerts, K.; Kubo, K. J. Med. Chem. 2010, 53, 3517–3531. doi:10.1021/jm901699j
Return to citation in text: [1] -
Marcinkowska, M.; Bucki, A.; Sniecikowska, J.; Zagórska, A.; Fajkis-Zajączkowska, N.; Siwek, A.; Gluch-Lutwin, M.; Żmudzki, P.; Jastrzebska-Wiesek, M.; Partyka, A.; Wesołowska, A.; Abram, M.; Przejczowska-Pomierny, K.; Cios, A.; Wyska, E.; Mika, K.; Kotańska, M.; Mierzejewski, P.; Kolaczkowski, M. J. Med. Chem. 2021, 64, 12603–12629. doi:10.1021/acs.jmedchem.1c00497
Return to citation in text: [1] -
Mannich, C.; Krösche, W. Arch. Pharm. (Weinheim, Ger.) 1912, 250, 647–667. doi:10.1002/ardp.19122500151
Return to citation in text: [1] -
Subramaniapillai, S. G. J. Chem. Sci. 2013, 125, 467–482. doi:10.1007/s12039-013-0405-y
Return to citation in text: [1] -
Kalck, P.; Urrutigoïty, M. Chem. Rev. 2018, 118, 3833–3861. doi:10.1021/acs.chemrev.7b00667
Return to citation in text: [1] -
Remeur, C.; Kelly, C. B.; Patel, N. R.; Molander, G. A. ACS Catal. 2017, 7, 6065–6069. doi:10.1021/acscatal.7b01973
Return to citation in text: [1] -
Yang, J.; Chen, S.; Zhou, H.; Wu, C.; Ma, B.; Xiao, J. Org. Lett. 2018, 20, 6774–6779. doi:10.1021/acs.orglett.8b02892
Return to citation in text: [1] -
Taniguchi, Y.; Horie, S.; Takaki, K.; Fujiwara, Y. J. Organomet. Chem. 1995, 504, 137–141. doi:10.1016/0022-328x(95)05605-o
Return to citation in text: [1] -
Shi, A.; Xie, P.; Wang, Y.; Qiu, Y. Nat. Commun. 2025, 16, 2322. doi:10.1038/s41467-025-57567-2
Return to citation in text: [1] -
Miyazawa, K.; Koike, T.; Akita, M. Adv. Synth. Catal. 2014, 356, 2749–2755. doi:10.1002/adsc.201400556
Return to citation in text: [1] -
Kaiser, D.; Tona, V.; Gonçalves, C. R.; Shaaban, S.; Oppedisano, A.; Maulide, N. Angew. Chem., Int. Ed. 2019, 58, 14639–14643. doi:10.1002/anie.201906910
Return to citation in text: [1] -
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y
Return to citation in text: [1] -
Venditto, N. J.; Boerth, J. A. Org. Lett. 2023, 25, 3429–3434. doi:10.1021/acs.orglett.3c00991
Return to citation in text: [1] -
Hurst, T. E.; Gorman, R. M.; Drouhin, P.; Perry, A.; Taylor, R. J. K. Chem. – Eur. J. 2014, 20, 14063–14073. doi:10.1002/chem.201403917
Return to citation in text: [1] -
Liang, J.; Chen, J.; Liu, J.; Li, L.; Zhang, H. Chem. Commun. 2010, 46, 3666–3668. doi:10.1039/c001465f
Return to citation in text: [1] -
Beveridge, R. E.; Hu, Y.; Gregoire, B.; Batey, R. A. J. Org. Chem. 2020, 85, 8447–8461. doi:10.1021/acs.joc.0c00781
Return to citation in text: [1] -
Huang, L.; Cao, D.; Pan, J.; Fang, L.; Wang, Q.; Chen, D.; Yang, J. Adv. Synth. Catal. 2020, 362, 5653–5657. doi:10.1002/adsc.202000974
Return to citation in text: [1] -
Smorodina, A. A.; Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. Tetrahedron Lett. 2023, 133, 154830. doi:10.1016/j.tetlet.2023.154830
Return to citation in text: [1] -
Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. Org. Chem. Front. 2025, 12, 1927–1935. doi:10.1039/d4qo02259a
Return to citation in text: [1] [2] [3] -
Curti, C.; Laget, M.; Carle, A. O.; Gellis, A.; Vanelle, P. Eur. J. Med. Chem. 2007, 42, 880–884. doi:10.1016/j.ejmech.2006.12.015
Return to citation in text: [1] -
Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. J. Org. Chem. 2017, 82, 12827–12833. doi:10.1021/acs.joc.7b02193
Return to citation in text: [1] -
Gorbunova, E. V.; Moshkin, V. S.; Fedorenko, V. D.; Khardina, P. A.; Buev, E. M.; Sosnovskikh, V. Y. Tetrahedron Lett. 2022, 112, 154227. doi:10.1016/j.tetlet.2022.154227
Return to citation in text: [1] -
Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1981, 46, 4327–4331. doi:10.1021/jo00335a001
Return to citation in text: [1] -
Malatesti, N.; Boa, A. N.; Clark, S.; Westwood, R. Tetrahedron Lett. 2006, 47, 5139–5142. doi:10.1016/j.tetlet.2006.05.064
Return to citation in text: [1] -
Lakshmi, N. V.; Thirumurugan, P.; Jayakumar, C.; Perumal, P. T. Synlett 2010, 955–961. doi:10.1055/s-0029-1219550
Return to citation in text: [1] -
Sankar, U.; Surya Kumar, C. V.; Subramanian, V.; Balasubramanian, K. K.; Mahalakshimi, S. J. Org. Chem. 2016, 81, 2340–2354. doi:10.1021/acs.joc.5b02845
Return to citation in text: [1] -
Zaika, Y. O.; Vashchenko, B. V.; Sosunovych, B. S.; Khutorianskyi, A. V.; Yasman, P.; Ogurok, V. M.; Brovarets, V. S.; Grygorenko, O. O. Eur. J. Org. Chem. 2025, 28, e202500093. doi:10.1002/ejoc.202500093
Return to citation in text: [1] -
Gorman, R. M.; Hurst, T. E.; Petersen, W. F.; Taylor, R. J. K. Tetrahedron 2019, 75, 130711. doi:10.1016/j.tet.2019.130711
Return to citation in text: [1] -
Bordwell, F. G.; McKellin, W. H. J. Am. Chem. Soc. 1951, 73, 2251–2253. doi:10.1021/ja01149a096
Return to citation in text: [1] -
Akgün, E.; Mahmood, K.; Mathis, C. A. J. Chem. Soc., Chem. Commun. 1994, 761–762. doi:10.1039/c39940000761
Return to citation in text: [1] -
Fong, H. O.; Hardstaff, W. R.; Kay, D. G.; Langler, R. F.; Morse, R. H.; Sandoval, D.-N. Can. J. Chem. 1979, 57, 1206–1213. doi:10.1139/v79-196
Return to citation in text: [1]
| 43. | Bordwell, F. G.; McKellin, W. H. J. Am. Chem. Soc. 1951, 73, 2251–2253. doi:10.1021/ja01149a096 |
| 44. | Akgün, E.; Mahmood, K.; Mathis, C. A. J. Chem. Soc., Chem. Commun. 1994, 761–762. doi:10.1039/c39940000761 |
| 45. | Fong, H. O.; Hardstaff, W. R.; Kay, D. G.; Langler, R. F.; Morse, R. H.; Sandoval, D.-N. Can. J. Chem. 1979, 57, 1206–1213. doi:10.1139/v79-196 |
| 1. | Supuran, C. T.; Casini, A.; Scozzafava, A. Med. Res. Rev. 2003, 23, 535–558. doi:10.1002/med.10047 |
| 2. | Ahmadi, R.; Emami, S. Eur. J. Med. Chem. 2022, 234, 114255. doi:10.1016/j.ejmech.2022.114255 |
| 3. | Huang, H.-L.; Shi, Y.-Q.; Ning, J.-X.; Li, S.; Song, D.-T.; Gao, F.; Ma, N. Molecules 2024, 29, 5868. doi:10.3390/molecules29245868 |
| 8. | Tucker, H.; Crook, J. W.; Chesterson, G. J. J. Med. Chem. 1988, 31, 954–959. doi:10.1021/jm00400a011 |
| 9. | Parent, E. E.; Dence, C. S.; Jenks, C.; Sharp, T. L.; Welch, M. J.; Katzenellenbogen, J. A. J. Med. Chem. 2007, 50, 1028–1040. doi:10.1021/jm060847r |
| 24. | Miyazawa, K.; Koike, T.; Akita, M. Adv. Synth. Catal. 2014, 356, 2749–2755. doi:10.1002/adsc.201400556 |
| 25. | Kaiser, D.; Tona, V.; Gonçalves, C. R.; Shaaban, S.; Oppedisano, A.; Maulide, N. Angew. Chem., Int. Ed. 2019, 58, 14639–14643. doi:10.1002/anie.201906910 |
| 26. | Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y |
| 27. | Venditto, N. J.; Boerth, J. A. Org. Lett. 2023, 25, 3429–3434. doi:10.1021/acs.orglett.3c00991 |
| 6. | Franca, J. R.; Foureaux, G.; Fuscaldi, L. L.; Ribeiro, T. G.; Castilho, R. O.; Yoshida, M. I.; Cardoso, V. N.; Fernandes, S. O. A.; Cronemberger, S.; Nogueira, J. C.; Ferreira, A. J.; Faraco, A. A. G. Int. J. Pharm. 2019, 570, 118662. doi:10.1016/j.ijpharm.2019.118662 |
| 21. | Yang, J.; Chen, S.; Zhou, H.; Wu, C.; Ma, B.; Xiao, J. Org. Lett. 2018, 20, 6774–6779. doi:10.1021/acs.orglett.8b02892 |
| 4. | Trost, B. M.; Kalnmals, C. A. Chem. – Eur. J. 2019, 25, 11193–11213. doi:10.1002/chem.201902019 |
| 5. | Nambo, M.; Maekawa, Y.; Crudden, C. M. ACS Catal. 2022, 12, 3013–3032. doi:10.1021/acscatal.1c05608 |
| 22. | Taniguchi, Y.; Horie, S.; Takaki, K.; Fujiwara, Y. J. Organomet. Chem. 1995, 504, 137–141. doi:10.1016/0022-328x(95)05605-o |
| 23. | Shi, A.; Xie, P.; Wang, Y.; Qiu, Y. Nat. Commun. 2025, 16, 2322. doi:10.1038/s41467-025-57567-2 |
| 15. | Fujimoto, T.; Imaeda, Y.; Konishi, N.; Hiroe, K.; Kawamura, M.; Textor, G. P.; Aertgeerts, K.; Kubo, K. J. Med. Chem. 2010, 53, 3517–3531. doi:10.1021/jm901699j |
| 17. | Mannich, C.; Krösche, W. Arch. Pharm. (Weinheim, Ger.) 1912, 250, 647–667. doi:10.1002/ardp.19122500151 |
| 18. | Subramaniapillai, S. G. J. Chem. Sci. 2013, 125, 467–482. doi:10.1007/s12039-013-0405-y |
| 19. | Kalck, P.; Urrutigoïty, M. Chem. Rev. 2018, 118, 3833–3861. doi:10.1021/acs.chemrev.7b00667 |
| 14. | Al-Riyami, L.; Pineda, M. A.; Rzepecka, J.; Huggan, J. K.; Khalaf, A. I.; Suckling, C. J.; Scott, F. J.; Rodgers, D. T.; Harnett, M. M.; Harnett, W. J. Med. Chem. 2013, 56, 9982–10002. doi:10.1021/jm401251p |
| 20. | Remeur, C.; Kelly, C. B.; Patel, N. R.; Molander, G. A. ACS Catal. 2017, 7, 6065–6069. doi:10.1021/acscatal.7b01973 |
| 13. | Aranapakam, V.; Davis, J. M.; Grosu, G. T.; Baker; Ellingboe, J.; Zask, A.; Levin, J. I.; Sandanayaka, V. P.; Du, M.; Skotnicki, J. S.; DiJoseph, J. F.; Sung, A.; Sharr, M. A.; Killar, L. M.; Walter, T.; Jin, G.; Cowling, R.; Tillett, J.; Zhao, W.; McDevitt, J.; Xu, Z. B. J. Med. Chem. 2003, 46, 2376–2396. doi:10.1021/jm0205550 |
| 10. | Zhang, H.; Zhao, X.; Yan, R.; Lin, W. Org. Lett. 2024, 26, 4251–4256. doi:10.1021/acs.orglett.4c01103 |
| 11. | Qian, Z.-M.; Wang, F.-Y.; Guan, Z.; He, Y.-H. Adv. Synth. Catal. 2024, 366, 3022–3028. doi:10.1002/adsc.202400432 |
| 12. | Griffiths, R. J.; Kong, W. C.; Richards, S. A.; Burley, G. A.; Willis, M. C.; Talbot, E. P. A. Chem. Sci. 2018, 9, 2295–2300. doi:10.1039/c7sc04900e |
| 16. | Marcinkowska, M.; Bucki, A.; Sniecikowska, J.; Zagórska, A.; Fajkis-Zajączkowska, N.; Siwek, A.; Gluch-Lutwin, M.; Żmudzki, P.; Jastrzebska-Wiesek, M.; Partyka, A.; Wesołowska, A.; Abram, M.; Przejczowska-Pomierny, K.; Cios, A.; Wyska, E.; Mika, K.; Kotańska, M.; Mierzejewski, P.; Kolaczkowski, M. J. Med. Chem. 2021, 64, 12603–12629. doi:10.1021/acs.jmedchem.1c00497 |
| 31. | Huang, L.; Cao, D.; Pan, J.; Fang, L.; Wang, Q.; Chen, D.; Yang, J. Adv. Synth. Catal. 2020, 362, 5653–5657. doi:10.1002/adsc.202000974 |
| 28. | Hurst, T. E.; Gorman, R. M.; Drouhin, P.; Perry, A.; Taylor, R. J. K. Chem. – Eur. J. 2014, 20, 14063–14073. doi:10.1002/chem.201403917 |
| 29. | Liang, J.; Chen, J.; Liu, J.; Li, L.; Zhang, H. Chem. Commun. 2010, 46, 3666–3668. doi:10.1039/c001465f |
| 30. | Beveridge, R. E.; Hu, Y.; Gregoire, B.; Batey, R. A. J. Org. Chem. 2020, 85, 8447–8461. doi:10.1021/acs.joc.0c00781 |
| 5. | Nambo, M.; Maekawa, Y.; Crudden, C. M. ACS Catal. 2022, 12, 3013–3032. doi:10.1021/acscatal.1c05608 |
| 42. | Gorman, R. M.; Hurst, T. E.; Petersen, W. F.; Taylor, R. J. K. Tetrahedron 2019, 75, 130711. doi:10.1016/j.tet.2019.130711 |
| 33. | Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. Org. Chem. Front. 2025, 12, 1927–1935. doi:10.1039/d4qo02259a |
| 37. | Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1981, 46, 4327–4331. doi:10.1021/jo00335a001 |
| 38. | Malatesti, N.; Boa, A. N.; Clark, S.; Westwood, R. Tetrahedron Lett. 2006, 47, 5139–5142. doi:10.1016/j.tetlet.2006.05.064 |
| 39. | Lakshmi, N. V.; Thirumurugan, P.; Jayakumar, C.; Perumal, P. T. Synlett 2010, 955–961. doi:10.1055/s-0029-1219550 |
| 40. | Sankar, U.; Surya Kumar, C. V.; Subramanian, V.; Balasubramanian, K. K.; Mahalakshimi, S. J. Org. Chem. 2016, 81, 2340–2354. doi:10.1021/acs.joc.5b02845 |
| 41. | Zaika, Y. O.; Vashchenko, B. V.; Sosunovych, B. S.; Khutorianskyi, A. V.; Yasman, P.; Ogurok, V. M.; Brovarets, V. S.; Grygorenko, O. O. Eur. J. Org. Chem. 2025, 28, e202500093. doi:10.1002/ejoc.202500093 |
| 4. | Trost, B. M.; Kalnmals, C. A. Chem. – Eur. J. 2019, 25, 11193–11213. doi:10.1002/chem.201902019 |
| 34. | Curti, C.; Laget, M.; Carle, A. O.; Gellis, A.; Vanelle, P. Eur. J. Med. Chem. 2007, 42, 880–884. doi:10.1016/j.ejmech.2006.12.015 |
| 35. | Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. J. Org. Chem. 2017, 82, 12827–12833. doi:10.1021/acs.joc.7b02193 |
| 36. | Gorbunova, E. V.; Moshkin, V. S.; Fedorenko, V. D.; Khardina, P. A.; Buev, E. M.; Sosnovskikh, V. Y. Tetrahedron Lett. 2022, 112, 154227. doi:10.1016/j.tetlet.2022.154227 |
| 32. | Smorodina, A. A.; Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. Tetrahedron Lett. 2023, 133, 154830. doi:10.1016/j.tetlet.2023.154830 |
| 33. | Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. Org. Chem. Front. 2025, 12, 1927–1935. doi:10.1039/d4qo02259a |
| 33. | Buev, E. M.; Moshkin, V. S.; Sosnovskikh, V. Y. Org. Chem. Front. 2025, 12, 1927–1935. doi:10.1039/d4qo02259a |
© 2026 Buev et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.