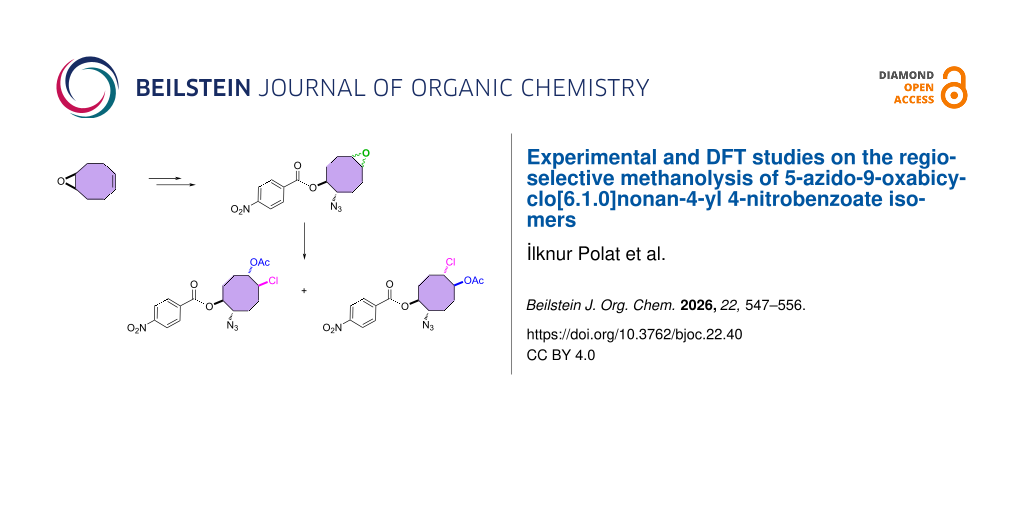
Abstract
The regioselective methanolysis of new azido-4-nitrobenzoate epoxycyclooctane isomers and the characterization of the resulting products are described herein. Firstly, treatment of key compound 8-azidocyclooct-4-en-1-ol with 4-nitrobenzoyl chloride followed by an epoxidation reaction and then methanolysis of the epoxide ring and acetylation resulted in the formation of two corresponding chloro-acetate isomers. The structure of one of the chloro-acetate isomers was determined via crystallographic analysis and the other by 1D and 2D NMR spectroscopy. DFT computations confirm the regioselectivity of the methanolysis process, highlighting its precision and efficiency.

Graphical Abstract
Introduction
Organic azides are very important precursors for the preparation and synthesis of various nitrogen-containing compounds, as well as triazoles and aminocyclitols, which have numerous applications in industry, medicine, and pharmacy [1-5]. They are found in many biologically active compounds. For example, 3’-azido-3’-deoxythymidine (zidovudine, 1) is used in treatment of AIDS, while azapride (2), azidamphenicol (3), and azidomorphine (4) are utilized as pharmaceuticals (Figure 1) [1,6-9]. Additionally, recent studies demonstrated that nonglycosyl azides hamper glycan acceptance by carbohydrate-processing enzymes [10,11]. Moreover, azido sugars are used directly in cell biology studies. Therefore, synthetic methodologies for the preparation of this class of compounds are of considerable interest.
![[1860-5397-22-40-1]](/bjoc/content/figures/1860-5397-22-40-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some important bioactive molecules with an azide group.
Figure 1: Some important bioactive molecules with an azide group.
Azides have traditionally been used as protected primary amine equivalents. Therefore, in our previous studies, we employed azides containing eight-membered rings as precursors in the synthesis of C8-aminocyclitols [12-19].
The ring-opening reactions of bicyclic epoxides are of significant importance and scientific interest due to their high ring strain, their strategic role in the construction of complex molecular architectures, and their utility in the synthesis of natural products, pharmaceutical compounds, and chiral ligands. The Lewis acid-catalyzed ring-opening reaction of cyclohexene oxide by MeZH (Z = O, S, and NH) is well known [20]. However, studies on large-sized bicyclic epoxides remain relatively limited in the published literature. In our previous study, we performed the ring-opening reaction of an azidoepoxide containing an eight-membered ring with HCl(g) in MeOH by regioselectivity [12].
More recently we have directed our interest towards developing an efficient method for the synthesis of new C8-azides. In the present paper, we report the first synthesis of 5-azido-9-oxabicyclo[6.1.0]nonan-4-yl 4-nitrobenzoate isomers starting from cis,cis-1,5-cyclooctadiene.
Results and Discussion
For the synthesis of the hitherto unknown key compound 8-azidocyclooct-4-en-1-yl 4-nitrobenzoate (8), we commenced by obtaining the known 5,6-epoxycyclooctene (6), which was prepared by epoxidation of cis,cis-1,5-cyclooctadiene (5) with m-CPBA [21,22]. Subsequently, the 5,6-epoxycyclooctene (6) was reacted with NaN3 and NH4Cl in EtOH/H2O to give azidol 7 in 92% yield [23] (Scheme 1). Azidol 7 was then converted into the corresponding azido nitrobenzoate 8 with p-nitrobenzoyl chloride (p-NBC) in pyridine and 4-(dimethylamino)pyridine (DMAP) in 95% yield. Treatment of azido nitrobenzoate 8 with m-CPBA gave a mixture of isomeric epoxides 9a and 9b with a combined yield of 97%. The formation and ratio of these isomers were determined by NMR spectroscopy, showing a 63:37 ratio (1H NMR) of 9a and 9b, respectively, but all attempts to separate the isomeric epoxides 9a and 9b by column chromatography or crystallization failed.
![[1860-5397-22-40-i1]](/bjoc/content/inline/1860-5397-22-40-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Epoxidation of azido nitrobenzoate 8.
Scheme 1: Epoxidation of azido nitrobenzoate 8.
Eight-membered rings are known to preferentially adopt boat-chair conformations, which minimize torsional and transannular strain [24]. To further rationalize the observed regioselectivity, steric drawings of epoxides 9a and 9b were constructed using a bathtub-like representation of the cyclooctane ring (Table 1). For epoxide 9a, the majority of dihedral angles fall within the ±60–80° range, consistent with a regular boat-chair geometry. The limited deviation from ideal gauche values and the absence of severely over-twisted dihedrals indicate a relatively low-strain conformation. In contrast, epoxide 9b exhibits a more distorted dihedral distribution, including a strongly twisted dihedral (≈90°) together with unevenly distributed gauche and near-planar torsions. This pattern is consistent with a less favourable, distorted boat-chair (or twist-boat-chair-like) conformation, associated with increased torsional strain, thereby disfavouring formation of 9b relative to 9a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-22-40-i4.png?max-width=637&scale=1.0)
Next, we turned our attention to the methanolysis of the mixture of isomeric epoxides 9. A ring-opening reaction of the epoxides 9 with HCl(g) in MeOH resulted in the formation of chlorohydrin-azidobenzoate isomers 10 and 11 in a total yield of 96% (Scheme 2). Then, for full characterization of the structures of the obtained products, the mixture of chlorohydrins was converted into the corresponding acetates 10 and 11 using AcCl in CH2Cl2. The reaction mixture was chromatographed on a silica gel column with n-hexane/ethyl acetate 85:15 as eluent to give pure acetates 10 and 11 in 57% and 35% yields, respectively. The structure of compound 10 was unambiguously determined by single crystal X-ray analysis (Figure 2) [25]. X-ray analysis of azido compound 10 confirmed the trans configuration of the azide group with chloride and the cis configuration with the acetate.
![[1860-5397-22-40-i2]](/bjoc/content/inline/1860-5397-22-40-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Methanolysis of the mixture of isomeric epoxides 9a and 9b.
Scheme 2: Methanolysis of the mixture of isomeric epoxides 9a and 9b.
![[1860-5397-22-40-2]](/bjoc/content/figures/1860-5397-22-40-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The X-ray crystal structure of 10.
Figure 2: The X-ray crystal structure of 10.
Our suggested mechanism for the ring-opening of the epoxides 9 with HCl(g) in MeOH proceeded as described in Scheme 3. As shown, the syn-face of the epoxide isomers 9a and 9b with the benzoate in 9a and the azide in 9b is more blocked than the anti-face. Therefore, the sterically hindering effect of these groups is observed in the syn-face. We assume that the chloride anion prefers to first attack the epoxide ring of intermediate 12 with the anti-face of the benzoate to give compound 14. Similarly, the chloride anion attacks by SN2-type the epoxide ring of intermediate 15 to give the sole product 16.
![[1860-5397-22-40-i3]](/bjoc/content/inline/1860-5397-22-40-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Suggested mechanism for the reaction of the isomeric epoxides 9a and 9b with HCl(g) in MeOH.
Scheme 3: Suggested mechanism for the reaction of the isomeric epoxides 9a and 9b with HCl(g) in MeOH.
The structure of acetate 11 was investigated using 1D (1H and 13C) and 2D (COSY, NOESY, NOE-diff, and HMQC) NMR spectroscopic data. However, the uncertainty or weakness of the cross peaks in NOESY and NOE signals indicated that compound 11 exhibits significant conformational flexibility. The cyclooctane ring has increased flexibility, so the configuration assignment of 11 is difficult to determine with the help of the NOESY experiments or the other 2D NMR conducted. Compound 11 is not a single crystal, so the determination by X-ray analysis of its structure was not possible. We strongly assume that the structure of 11 is based on the methanolysis mechanism of the epoxide 9a (Scheme 3).
In order to elucidate the formation mechanisms of the products yielded from the methanolysis of isomeric epoxides 9, we performed a series of density functional theory (DFT) computations using the software Gaussian 16 [26]. For this, geometry optimizations and harmonic vibrational frequency computations for the structures considered were carried out with DFT at the M06-2X hybrid functional [27] augmented by Grimme’s dispersion correction DFT-D3 to improve the description of long-range dispersion interactions [28]. For this purpose, a Pople-type polarized triple-ζ split valence basis set with diffuse functions on all atoms, 6-311G++(d,p) [29-31], and SMD [32] model (methanol (ε = 32.613)) were employed. Initial geometries were pre-optimized using MM2 (ChemDraw 3D) prior to DFT optimization to account for conformational flexibility. The 3D molecular structures were visualized using the open-source software cheMVP.exe.
DFT computations were performed to gain insights into the mechanism underlying regioselectivity for the methanolysis of the isomeric epoxides 9 (Figure 3). Detailed mechanistic routes involving all possible intermediates responsible for the regioselectivity step were constructed and the free energy barriers for each elementary reaction step along these routes were computed. The acid-mediated ring-opening reaction in the isomeric epoxides 9, initiated by protonation, can follow two possible pathways – either along the intermediate 12 or along the intermediate 15. There is a possibility of two products being formed in both pathways. Free energy computations indicate that the intermediate 12 is thermodynamically more stable by 4.9 kcal mol−1 compared to intermediate 15. The nucleophilic attack by chloride ion bifurcates into two paths, namely C1- (route a) and C2-attacks (route b) for intermediates 12 and 15. Halohydrin 14 is yielded with a C1-attack on intermediate 12, while 13 is yielded with a C2-attack. For the formation of halohydrin 14, 12 → 14, the computed reaction and activation free energies are −12.9 and 19.0 kcal mol−1, respectively, while for the 12 → 13 conversion, the computed reaction and activation free energies are −12.5 and 23.7 kcal mol−1, respectively. Halohydrin 16 is yielded by a C2-attack on 15, whereas a C1-attack yields 17. For the formation of halohydrin 16, 15 → 16, the computed reaction and activation free energies are −16.2 and 11.2 kcal mol−1, respectively, while for 15 → 17 conversion, the computed reaction and activation free energies are −17.9 and 18.1 kcal mol−1, respectively. The DFT results are rationally explained and supported our experimental observations, indicating that the methanolysis of both epoxides 9a and 9b preferentially follows the kinetically favourable pathway.
![[1860-5397-22-40-3]](/bjoc/content/figures/1860-5397-22-40-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Relative free energy profile for the methanolysis of the isomeric epoxides 9.
Figure 3: Relative free energy profile for the methanolysis of the isomeric epoxides 9.
The conformational preferences of the cyclooctane ring were analyzed based on the eight consecutive dihedral angles (τ1–τ8, °), which are summarized in Table 2, following the classification proposed by Pakes and co-workers [24]. TS1 displays a predominantly boat-chair geometry, with only one over-twisted dihedral angle (τ3 = 94°), indicating a moderately distorted boat-chair conformation. In contrast, TS2 exhibits a characteristic twist-boat-chair arrangement, evidenced by the co-existence of a strongly over-twisted segment (τ2 = 101.8°) and a nearly planar dihedral (τ6 = 4.0°). A similar trend is observed for TS3 and TS4. TS3 retains a largely boat-chair-like geometry, however, the presence of a single over-twisted dihedral (τ7 = 93.3°) indicates the onset of conformational distortion. By contrast, TS4 exhibits multiple over-twisted dihedrals (τ2 = 106.1° and τ7 = 104.0°, τ3= −88.2°) together with a nearly planar dihedral (τ5 = −15.5°), unambiguously identifying this structure as a strongly distorted twist-boat-chair conformation. Overall, these results demonstrate that progression along the reaction coordinate is accompanied by increasing torsional strain within the cyclooctane ring, which correlates well with the computed activation barriers and the divergence of the competing reaction pathways. In this context, despite the presence of a hydrogen-bonding interaction in TS2, TS1 is lower in energy due to a more favourable cyclooctane conformation. Notably, the relatively small energy difference between TS1 and TS2 (4.7 kcal/mol), compared to that between TS3 and TS4 (6.9 kcal/mol), can be attributed to hydrogen-bond stabilization in TS2, indicating that conformational effects play a more dominant role than hydrogen bonding in determining transition-state stability.
Table 2: Dihedral angles (τ, °) in the optimized transition states.
![[Graphic 2]](/bjoc/content/inline/1860-5397-22-40-i5.png?max-width=637&scale=1.0)
|
|||||||||
| TS | τ1 | τ2 | τ3 | τ4 | τ5 | τ6 | τ7 | τ8 | conformer |
| TS1 | −67.8 | −48.3 | 94.0 | −30.9 | 22.6 | −74.7 | 40.0 | 68.8 | boat-chair |
| TS2 | −37.9 | 101.8 | −62.9 | 22.0 | −28.7 | 4.0 | 70.9 | −66.3 | twist-boat-chair |
| TS3 | −67.0 | 62.5 | 44.4 | −76.6 | 22.5 | −32.3 | 93.3 | −45.3 | boat-chair |
| TS4 | −61.9 | 106.1 | −88.2 | 62.8 | −15.5 | −67.2 | 104.0 | −27.2 | twist-boat-chair |
Mulliken charge analysis for the protonated epoxide intermediates 12 and 15, derived from epoxides 9a and 9b, respectively, reveals that the charge differences between the epoxide carbons are not pronounced for either diastereomer, indicating that electronic effects alone are insufficient to explain the observed regioselectivity (Figure 4). In the protonated intermediate 12 derived from epoxide 9a, the carbon atom preferentially attacked by chloride bears a relatively more positive charge, which is consistent with an electronically favoured nucleophilic attack. In contrast, for the protonated intermediate 15 derived from epoxide 9b, the more positively charged epoxide carbon is sterically shielded by the neighbouring ester carbonyl group. As a result, nucleophilic attack occurs at the sterically more accessible, albeit less positively charged, epoxide carbon. These observations indicate that the regioselectivity of epoxide 9a is governed by a combination of electronic and steric factors, while steric effects are more pronounced for epoxide 9b.
![[1860-5397-22-40-4]](/bjoc/content/figures/1860-5397-22-40-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Mulliken charge analysis of protonated epoxide intermediates 12 and 15.
Figure 4: Mulliken charge analysis of protonated epoxide intermediates 12 and 15.
Conclusion
We achieved the first synthesis of new cyclooctane-azide derivatives (8, 10, and 11) efficiently from commercially readily available cis,cis-1,5-cyclooctadiene. The key step, the ring opening of epoxide 9, proceeded smoothly by methanolysis with HCl(g) in MeOH. The regioselectivity of oxirane-ring opening in 9 was attributed to the conformational effects. The mechanism for the formation of compounds 10 and 11 was elucidated with DFT computations. TS1 and TS3 predominantly retain boat-chair-like geometries, whereas TS2 and especially TS4 adopt significantly distorted twist-boat-chair conformations. The associated torsional strain accounts for the increased activation barriers and favours regioselectivity. These novel compounds synthesized may be used as precursors to design pharmacological tools in the future.
Experimental
General information
Melting points are uncorrected. Infrared spectra were obtained from solution in 0.1 mm cells or KBr pellets on an FT-IR Mattson 1000 instrument. The 1H and 13C NMR spectra were recorded on 400 (100) MHz Varian or 400 (100) MHz Bruker spectrometers and are reported in δ units with SiMe4 as internal standard. HRMS spectra were obtained on a Bruker microTOF-Q or Agilent 6530 Accurate Mass Q-TOF instrument. Melting points were determined on a GallenKamp MPD 350. Column chromatography was performed on silica gel (60 mesh, Merck). TLC was carried out on Merck 0.2 mm silica gel 60 F254 analytical aluminium plates.
(1R*,8S*,Z)-9-Oxabicyclo[6.1.0]non-4-ene (6)
![[Graphic 3]](/bjoc/content/inline/1860-5397-22-40-i6.svg?max-width=637&scale=1.18182)
A magnetically stirred solution of cis,cis-1,5-cyclooctadiene (5, 1.00 g, 9.24 mmol) in 50 mL dichloromethane was cooled to 0 °C. m-CPBA (2.20 g, 77%, 9.82 mmol) and NaHCO3 (0.78 g, 9.29 mmol) were added to the solution and stirred for 1.5 hours. Subsequently, 50 mL of 1.5 M NaOH solution was added and the reaction mixture was stirred for 15 min at 0 °C, then extracted with dichloromethane (4 × 30 mL). The organic layer was dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography eluting with pure hexane to give as the first fraction monoepoxide 6 (1.09 g, 8.78 mmol, 95%, a colourless oil) and as the second bisepoxide (0.07 g, 0.5 mmol, 5%, a white solid). The spectroscopic data are in accordance with those reported in the literature [22].
(1S*,8S*,Z)-8-Azidocyclooct-4-en-1-ol (7)
![[Graphic 4]](/bjoc/content/inline/1860-5397-22-40-i7.svg?max-width=637&scale=1.18182)
Cyclooctene monoepoxide 6 (2.00 g, 16.11 mmol) was dissolved in a 3:2 mixture of ethanol/water (80 mL). NaN3 (6.28 g, 96.60 mmol) and NH4Cl (1.72 g, 32.16 mmol) were added and the reaction mixture was refluxed. The progress was monitored by TLC and the reaction was completed in 36 hours. The solvent was removed by rotary evaporation, 30 mL water was added to the residue and the mixture was extracted with ethyl acetate (4 × 25 mL). The combined organic phases were dried over Na2SO4 and concentrated in vacuo. Purification by silica gel column chromatography eluting with EtOAc/hexane 5:95 provided azidol 7 [23] as slightly yellow oil (2.47 g, 14.77 mmol, 92%, lit. [23] 71%). 1H NMR (400 MHz, CDCl3) δ 5.69–5.61 (m, 1H), 5.60–5.52 (m, 1H), 3.79–3.65 (m, 2H), 2.56–2.36 (m, 2H), 2.27–2.09 (m, 4H), 1.81–1.67 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 130.3, 127.5, 73.1, 66.8, 32.4, 29.7, 23.5, 23.1.
(1S*,8S*,Z)-8-Azidocyclooct-4-en-1-yl 4-nitrobenzoate (8)
![[Graphic 5]](/bjoc/content/inline/1860-5397-22-40-i8.svg?max-width=637&scale=1.18182)
The azidol 7 (0.50 g, 2.99 mmol) was dissolved in 9 mL of anhydrous pyridine and the solution was cooled to 0 °C. p-Nitrobenzoyl chloride (1.11 g, 5.98 mmol) and 4-(dimethylamino)pyridine (DMAP, 2 mg) were added and the mixture was stirred at room temperature for 24 hours. Then, the solution was cooled to 0 °C and 100 mL of 2 M HCl added and stirred for 5 min. The mixture was extracted with ethyl acetate (4 × 30 mL). The organic phase was washed with saturated NaHCO3 (2 × 25 mL) and water (2 × 30 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by chromatography on a silica gel column eluting with EtOAc/hexane 20:80 to give pure azido benzoate 8 (0.90 g, 2.85 mmol, 95%). The azido benzoate 8 was recrystallized from EtOAc/hexane to give slightly yellow crystals; mp 60–62 °C. 1H NMR (400 MHz, CDCl3) δ 8.29–8.19 (m, 4H, aromatic), 5.72–5.59 (m, 2H, H-4 and H-5), 5.36–5.27 (m, 1H, H-1), 3.96 (td, J = 9.2, 3.7 Hz, 1H, H-8), 2.60–1.71 (series of m, 8H, CH2); 13C NMR (100 MHz, CDCl3) δ 163.9, 150.8, 135.7, 131.1, 129.5, 128.3, 123.8, 77.7, 64.1, 30.7, 30.3, 23.8, 23.3; IR (cm−1): 3013, 2940, 2621, 2515, 2099, 1725, 1527, 1272, 1117, 1103; HRESIMS (m/z): [M+ + Na] calcd for C15H16N4O4, 339.1069; found, 339.1063.
Epoxidation of the azidobenzoate 8 with m-CPBA
![[Graphic 6]](/bjoc/content/inline/1860-5397-22-40-i9.svg?max-width=637&scale=1.18182)
The azidobenzoate 8 (1.00 g, 3.16 mmol) was dissolved in CH2Cl2 (70 mL) and cooled to 0 °C. m-CPBA (77%, 0.73 g, 3.26 mmol) and NaHCO3 (0.282 g, 3.36 mmol) were added and the mixture was stirred at room temperature for 36 hours. Then, the solution was cooled to 0 °C and 100 mL of 3 M NaOH added, and stirring continued for 40 min. The mixture was extracted with CH2Cl2 (4 × 30 mL). The combined organic extracts were washed with saturated aqueous NaCl (20 mL) and then dried (Na2SO4). Evaporation of solvent gave the mixture of azidobenzoate epoxides 9a and 9b (1.02 g, 3.07 mmol, 97%), as light brown solid. The formation and ratio of these isomers was determined by NMR spectroscopy, in a 63:37 ratio (1H NMR), but all attempts to separate the isomeric epoxides 9a and 9b by column chromatography or crystallization failed.
Acetylation and methanolysis of the mixture of isomeric azidobenzoate epoxides 9a and 9b with HCl(g) in MeOH
![[Graphic 7]](/bjoc/content/inline/1860-5397-22-40-i10.svg?max-width=637&scale=1.18182)
In a similar manner as described in the literature [13], a magnetically stirred solution of the mixture of isomeric epoxides 9a and 9b (1.00 g, 3.01 mmol) in 5 mL absolute methanol was cooled to 0 °C. Then, 20 mL absolute methanol containing 10% HCl gas was added and the reaction mixture stirred at 0 °C for 1 hour. The solvent was removed under reduced pressure to obtain the mixture of chlorohydrin-azidobenzoates (1.07 g, 2.90 mmol, 96%). This crude product was dissolved in dichloromethane at 0 °C, and acetyl chloride (0.46 mL, 6.38 mmol) was added dropwise. After 24 hours, evaporation of the solvent under reduced pressure gave the mixture of azidobenzoate chloro-acetates 10 and 11 (1.11 g, 2.70 mmol, total yield 93%). Chromatography of the mixture on a silica gel column eluting with EtOAc/hexane 15:85 gave as the first fraction compound 10 (675 mg, 57%) and as the second fraction compound 11 (417 mg, 35%). Compound 10 was recrystallized from dichloromethane/hexane at room temperature to obtain colourless crystals, mp: 101–102 °C. Compound 11 was recrystallized from the same solvent mixture at 0 °C and obtained as slightly yellow crystals, mp: 117–119 °C. (1S*,2S*,5S*,6S*)-6-Acetoxy-2-azido-5-chlorocyclooctyl 4-nitrobenzoate (10): 1H NMR (400 MHz, CDCl3) δ 8.34–8.22 (m, aromatic, 4H), 5.40–5.33 (m, 1H, H-1), 5.22–5.14 (m, 1H, H-6), 4.25–4.19 (m, 1H, H-5), 3.97 (ddd, J = 9.1, 6.0, 3.4 Hz, 1H, H-2), 2.36–1.90 (series of m, 8H, CH2), 2.16 (s, 3H, OAc); 13C NMR (100 MHz, CDCl3) δ 170.2, 164.0, 150.9, 135.3, 131.1, 123.9, 76.9, 75.8, 64.0, 61.5, 26.7, 25.0, 24.5, 21.2; IR: 2951, 2621, 2102, 1772, 1727, 1528, 1272, 1237, 1020, 719; HRESIMS (m/z): [M+ + Na] calcd for C17H19ClN4O6, 433.0891; found, 433.0884. (1S*,2S*,5S*,6S*)-5-Acetoxy-2-azido-6-chlorocyclooctyl 4-nitrobenzoate (11): 1H NMR (400 MHz, CDCl3) δ 8.36–8.24 (m, 4H, aromatic), 5.23–5.09 (m, 1H, H-1 and H-5), 4.20 (td, J = 8.2, 3.4 Hz, 1H, H-6), 3.91 (td, J = 9.3, 2.9 Hz, 1H, H-2), 2.44–1.86 (series of m, 8H, CH2), 2.14 (s, 3H, OAc); 13C NMR (100 MHz, CDCl3) δ 170.0, 163.9, 151.0, 135.2, 131.1, 123.9, 76.9, 75.1, 64.0, 61.4, 29.1, 27.1, 27.0, 25.8, 21.2; IR: 2952, 2515, 2103, 1947, 1728, 1529, 1271, 1239, 1015, 720; HRESIMS (m/z): [M+ + Na] calcd for C17H19ClN4O6, 433.0891; found, 433.0885.
Supporting Information
| Supporting Information File 1: Experimental, 1H and 13C NMR spectra for all new compounds, as well as selected 2D NMR spectra and crystallographic data for compound 10 are provided. Optimized geometries of the transition states with selected interatomic distances and cartesian coordinates for computed structures. | ||
| Format: PDF | Size: 2.2 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Bräse, S.; Banert, K., Eds. Organic Azides: Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. doi:10.1002/9780470682517
Return to citation in text: [1] [2] -
Carlson, A. S.; Topczewski, J. J. Org. Biomol. Chem. 2019, 17, 4406–4429. doi:10.1039/c8ob03178a
Return to citation in text: [1] -
Salamci, E. Tetrahedron Lett. 2020, 61, 151728. doi:10.1016/j.tetlet.2020.151728
Return to citation in text: [1] -
Salamci, E. Tetrahedron 2026, 189, 134988. doi:10.1016/j.tet.2025.134988
Return to citation in text: [1] -
Delgado, A. Eur. J. Org. Chem. 2008, 3893–3906. doi:10.1002/ejoc.200800238
Return to citation in text: [1] -
Lin, T.-S.; Prusoff, W. H. J. Med. Chem. 1978, 21, 106–109. doi:10.1021/jm00199a019
Return to citation in text: [1] -
Lowe-Ma, C. K.; Nissan, R. A.; Wilson, W. S. J. Org. Chem. 1990, 55, 3755–3761. doi:10.1021/jo00299a014
Return to citation in text: [1] -
Zhou, Y.; Murphy, P. V. Org. Lett. 2008, 10, 3777–3780. doi:10.1021/ol8014495
Return to citation in text: [1] -
Dürüst, Y.; Karakuş, H.; Kaiser, M.; Tasdemir, D. Eur. J. Med. Chem. 2012, 48, 296–304. doi:10.1016/j.ejmech.2011.12.028
Return to citation in text: [1] -
Hore, R.; Bit, K.; Pan, R.; Halder, T.; Das, S.; Sett, S.; Bera, T.; Subba, A.; Maity, J. ACS Omega 2025, 10, 42999–43011. doi:10.1021/acsomega.5c05437
Return to citation in text: [1] -
Liu, F.; Chen, H.-M.; Armstrong, Z.; Withers, S. G. ACS Cent. Sci. 2022, 8, 656–662. doi:10.1021/acscentsci.1c01172
Return to citation in text: [1] -
Karavaizoglu, U. N.; Salamci, E. New J. Chem. 2020, 44, 17976–17983. doi:10.1039/d0nj02697b
Return to citation in text: [1] [2] -
Salamci, E.; Zozik, Y. Beilstein J. Org. Chem. 2021, 17, 705–710. doi:10.3762/bjoc.17.59
Return to citation in text: [1] [2] -
Salamci, E.; Lafzi, A. K. Beilstein J. Org. Chem. 2022, 18, 1539–1543. doi:10.3762/bjoc.18.163
Return to citation in text: [1] -
Salamci, E.; Karavaizoglu, U. N. Tetrahedron Lett. 2023, 129, 154759. doi:10.1016/j.tetlet.2023.154759
Return to citation in text: [1] -
Zozik, Y.; Salamci, E.; Kilic, A. Tetrahedron Lett. 2017, 58, 4822–4826. doi:10.1016/j.tetlet.2017.11.014
Return to citation in text: [1] -
Ecer, K.; Salamci, E. Tetrahedron 2014, 70, 8389–8396. doi:10.1016/j.tet.2014.08.060
Return to citation in text: [1] -
Salamci, E.; Ustabaş, R.; Çoruh, U.; Yavuz, M.; Vázquez-López, E. M. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o2401–o2402. doi:10.1107/s1600536806018204
Return to citation in text: [1] -
Salamci, E. Tetrahedron 2010, 66, 4010–4015. doi:10.1016/j.tet.2010.04.052
Return to citation in text: [1] -
Hansen, T.; Vermeeren, P.; Yoshisada, R.; Filippov, D. V.; van der Marel, G. A.; Codée, J. D. C.; Hamlin, T. A. J. Org. Chem. 2021, 86, 3565–3573. doi:10.1021/acs.joc.0c02955
Return to citation in text: [1] -
Crandall, J. K.; Banks, D. B.; Colyer, R. A.; Watkins, R. J.; Arrington, J. P. J. Org. Chem. 1968, 33, 423–425. doi:10.1021/jo01265a089
Return to citation in text: [1] -
Hillmyer, M. A.; Laredo, W. R.; Grubbs, R. H. Macromolecules 1995, 28, 6311–6316. doi:10.1021/ma00122a043
Return to citation in text: [1] [2] -
Siegl, S. J.; Vázquez, A.; Dzijak, R.; Dračínský, M.; Galeta, J.; Rampmaier, R.; Klepetářová, B.; Vrabel, M. Chem. – Eur. J. 2018, 24, 2426–2432. doi:10.1002/chem.201705188
Return to citation in text: [1] [2] [3] -
Pakes, P. W.; Rounds, T. C.; Strauss, H. L. J. Phys. Chem. 1981, 85, 2469–2475. doi:10.1021/j150617a013
Return to citation in text: [1] [2] -
Crystallographic data for the structural analysis of compound 10 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as CCDC-2063912. These data are provided free of charge via the joint CCDC/FIZ Karlsruhe deposition service https://www.ccdc.cam.ac.uk/structures/.
Return to citation in text: [1] -
Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2019.
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344
Return to citation in text: [1] -
Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213–222. doi:10.1007/bf00533485
Return to citation in text: [1] -
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. doi:10.1063/1.438955
Return to citation in text: [1] -
McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980
Return to citation in text: [1] -
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n
Return to citation in text: [1]
| 23. | Siegl, S. J.; Vázquez, A.; Dzijak, R.; Dračínský, M.; Galeta, J.; Rampmaier, R.; Klepetářová, B.; Vrabel, M. Chem. – Eur. J. 2018, 24, 2426–2432. doi:10.1002/chem.201705188 |
| 22. | Hillmyer, M. A.; Laredo, W. R.; Grubbs, R. H. Macromolecules 1995, 28, 6311–6316. doi:10.1021/ma00122a043 |
| 23. | Siegl, S. J.; Vázquez, A.; Dzijak, R.; Dračínský, M.; Galeta, J.; Rampmaier, R.; Klepetářová, B.; Vrabel, M. Chem. – Eur. J. 2018, 24, 2426–2432. doi:10.1002/chem.201705188 |
| 1. | Bräse, S.; Banert, K., Eds. Organic Azides: Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. doi:10.1002/9780470682517 |
| 2. | Carlson, A. S.; Topczewski, J. J. Org. Biomol. Chem. 2019, 17, 4406–4429. doi:10.1039/c8ob03178a |
| 3. | Salamci, E. Tetrahedron Lett. 2020, 61, 151728. doi:10.1016/j.tetlet.2020.151728 |
| 4. | Salamci, E. Tetrahedron 2026, 189, 134988. doi:10.1016/j.tet.2025.134988 |
| 5. | Delgado, A. Eur. J. Org. Chem. 2008, 3893–3906. doi:10.1002/ejoc.200800238 |
| 20. | Hansen, T.; Vermeeren, P.; Yoshisada, R.; Filippov, D. V.; van der Marel, G. A.; Codée, J. D. C.; Hamlin, T. A. J. Org. Chem. 2021, 86, 3565–3573. doi:10.1021/acs.joc.0c02955 |
| 32. | Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n |
| 12. | Karavaizoglu, U. N.; Salamci, E. New J. Chem. 2020, 44, 17976–17983. doi:10.1039/d0nj02697b |
| 13. | Salamci, E.; Zozik, Y. Beilstein J. Org. Chem. 2021, 17, 705–710. doi:10.3762/bjoc.17.59 |
| 14. | Salamci, E.; Lafzi, A. K. Beilstein J. Org. Chem. 2022, 18, 1539–1543. doi:10.3762/bjoc.18.163 |
| 15. | Salamci, E.; Karavaizoglu, U. N. Tetrahedron Lett. 2023, 129, 154759. doi:10.1016/j.tetlet.2023.154759 |
| 16. | Zozik, Y.; Salamci, E.; Kilic, A. Tetrahedron Lett. 2017, 58, 4822–4826. doi:10.1016/j.tetlet.2017.11.014 |
| 17. | Ecer, K.; Salamci, E. Tetrahedron 2014, 70, 8389–8396. doi:10.1016/j.tet.2014.08.060 |
| 18. | Salamci, E.; Ustabaş, R.; Çoruh, U.; Yavuz, M.; Vázquez-López, E. M. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o2401–o2402. doi:10.1107/s1600536806018204 |
| 19. | Salamci, E. Tetrahedron 2010, 66, 4010–4015. doi:10.1016/j.tet.2010.04.052 |
| 24. | Pakes, P. W.; Rounds, T. C.; Strauss, H. L. J. Phys. Chem. 1981, 85, 2469–2475. doi:10.1021/j150617a013 |
| 10. | Hore, R.; Bit, K.; Pan, R.; Halder, T.; Das, S.; Sett, S.; Bera, T.; Subba, A.; Maity, J. ACS Omega 2025, 10, 42999–43011. doi:10.1021/acsomega.5c05437 |
| 11. | Liu, F.; Chen, H.-M.; Armstrong, Z.; Withers, S. G. ACS Cent. Sci. 2022, 8, 656–662. doi:10.1021/acscentsci.1c01172 |
| 28. | Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344 |
| 1. | Bräse, S.; Banert, K., Eds. Organic Azides: Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. doi:10.1002/9780470682517 |
| 6. | Lin, T.-S.; Prusoff, W. H. J. Med. Chem. 1978, 21, 106–109. doi:10.1021/jm00199a019 |
| 7. | Lowe-Ma, C. K.; Nissan, R. A.; Wilson, W. S. J. Org. Chem. 1990, 55, 3755–3761. doi:10.1021/jo00299a014 |
| 8. | Zhou, Y.; Murphy, P. V. Org. Lett. 2008, 10, 3777–3780. doi:10.1021/ol8014495 |
| 9. | Dürüst, Y.; Karakuş, H.; Kaiser, M.; Tasdemir, D. Eur. J. Med. Chem. 2012, 48, 296–304. doi:10.1016/j.ejmech.2011.12.028 |
| 29. | Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213–222. doi:10.1007/bf00533485 |
| 30. | Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. doi:10.1063/1.438955 |
| 31. | McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980 |
| 24. | Pakes, P. W.; Rounds, T. C.; Strauss, H. L. J. Phys. Chem. 1981, 85, 2469–2475. doi:10.1021/j150617a013 |
| 23. | Siegl, S. J.; Vázquez, A.; Dzijak, R.; Dračínský, M.; Galeta, J.; Rampmaier, R.; Klepetářová, B.; Vrabel, M. Chem. – Eur. J. 2018, 24, 2426–2432. doi:10.1002/chem.201705188 |
| 27. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 21. | Crandall, J. K.; Banks, D. B.; Colyer, R. A.; Watkins, R. J.; Arrington, J. P. J. Org. Chem. 1968, 33, 423–425. doi:10.1021/jo01265a089 |
| 22. | Hillmyer, M. A.; Laredo, W. R.; Grubbs, R. H. Macromolecules 1995, 28, 6311–6316. doi:10.1021/ma00122a043 |
| 13. | Salamci, E.; Zozik, Y. Beilstein J. Org. Chem. 2021, 17, 705–710. doi:10.3762/bjoc.17.59 |
| 12. | Karavaizoglu, U. N.; Salamci, E. New J. Chem. 2020, 44, 17976–17983. doi:10.1039/d0nj02697b |
| 25. | Crystallographic data for the structural analysis of compound 10 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as CCDC-2063912. These data are provided free of charge via the joint CCDC/FIZ Karlsruhe deposition service https://www.ccdc.cam.ac.uk/structures/. |
© 2026 Polat et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.