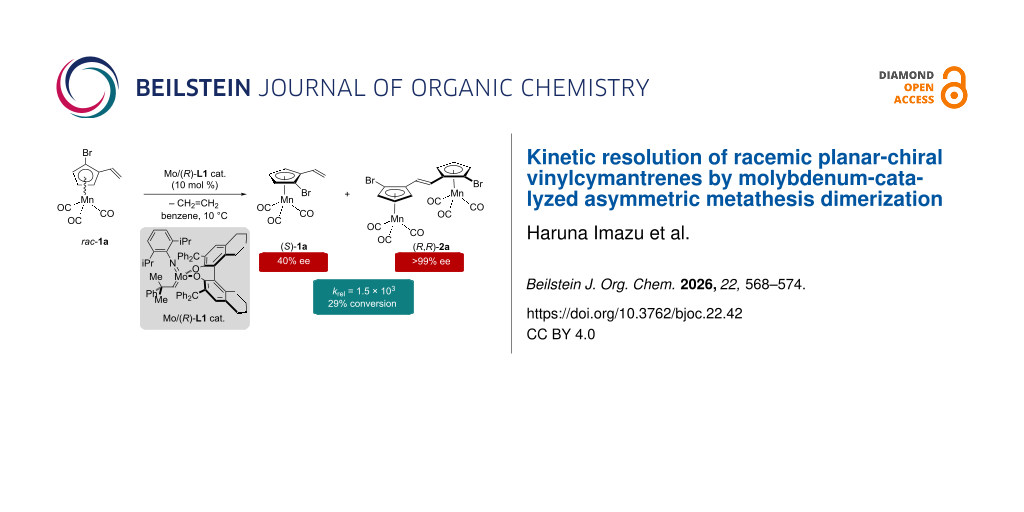
Abstract
Highly diastereo- and enantioselective kinetic resolution (KR) of a series of racemic planar-chiral 1-R-2-vinylcymantrenes (rac-1; R = Br, Me, I) was realized by an asymmetric metathesis dimerization (AMD) reaction catalyzed by a chiral molybdenum-alkylidene precatalyst. The Mo/(R)-L1 precatalyst promoted the AMD/KR reaction of rac-1a (R = Br) to give (E)-(R,R)-2a of 99% ee together with unreacted recovered (S)-1a of 45% ee at 37% conversion. The diastereoselectivity of this reaction was excellent with chiral-2a/meso-2a = 96:4 molar ratio, and the selectivity factor (krel) was calculated to be 754 based on a second-order equation. In all the three substrates examined, the dimerized products, chiral-2, were obtained in >98% ee thanks to the outstanding enantioselectivity.

Graphical Abstract
Introduction
The development of the well-defined molybdenum- [1,2] or ruthenium-alkylidene [3-5] catalysts has proven the olefin metathesis reaction to be a powerful tool in organic synthesis. Asymmetric extension is a recent trend in metathesis chemistry, and various chiral metal-alkylidene catalysts have been prepared over the past two decades [6-9].
Since 2002, our group and others have been interested in utilizing the olefin metathesis reaction for the modulation of various transition-metal complexes [10-13] thanks to the excellent tolerance of the Mo-/Ru-metathesis catalysts toward the organometallic substrates. The olefin metathesis protocols could be extended to the asymmetric synthesis of diverse planar-chiral transition metal complexes either by the kinetic resolution of the racemic substrates [14-16] or by the desymmetrization of the Cs-symmetric substrates [17-19] by the use of an appropriate chiral catalyst (see the drawing in Table 1 for the structures of the representative chiral molybdenum precatalysts used in this study) [20-23]. Planar-chiral transition-metal complexes have been demonstrated to be excellent chiral scaffolds in asymmetric synthesis, however, enantioselective synthesis of such planar-chiral complexes is still relatively undeveloped and remains as a challenging problem in asymmetric synthesis [24-29].
A new type of asymmetric olefin metathesis reaction, that is the asymmetric metathesis dimerization (AMD; Scheme 1), was developed by our group during 2022–23 [30,31]. In the previous reports, racemic planar-chiral vinylferrocene [30] or vinylphosphaferrocene derivatives [31] were employed as substrates for the AMD process. In the presence of an appropriate chiral Mo-alkylidene precatalyst, the highly diastereo- and enantioselective kinetic resolution (KR) of the racemic substrates was attained with the krel values ([reaction rate of the fast-reacting enantiomer]/[reaction rate of the slow-reacting enantiomer]; selectivity factor) of up to 96 for the vinylferrocenes and of over 1000 for the vinylphosphaferrocenes.
![[1860-5397-22-42-i1]](/bjoc/content/inline/1860-5397-22-42-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Asymmetric metathesis dimerization/kinetic resolution of racemic planar-chiral vinylferrocene/vinylphosphaferrocene.
Scheme 1: Asymmetric metathesis dimerization/kinetic resolution of racemic planar-chiral vinylferrocene/vinyl...
In this article, we would like to report the analogous asymmetric metathesis dimerization/kinetic resolution of a series of racemic planar-chiral vinylcymantrenes (rac-1a–c). It was found that the chiral molybdenum-alkylidene precatalysts Mo/(R)-L1 or Mo/(R)-L3 discriminate the two enantiomers in rac-1 efficiently to provide a mixture of the corresponding AMD product (S,S)-2 and unreacted antipodal substrate (R)-1 with the krel values of up to 1.5 × 103. The AMD/KR reactions of rac-1 were highly diastereoselective: under the optimized conditions, (S,S)-2 was the sole AMD product and the formation of the respective mesomeric stereoisomer was negligible. It should be noted that this work is a rare example of catalytic asymmetric synthesis of planar-chiral CpMn(I) half-sandwich complexes [19,32].
Results and Discussion
Preparation of racemic planar-chiral 2-substituted vinylcymantrene substrates rac-1a–c
A series of racemic planar-chiral vinylcymantrene substrates rac-1a–c were prepared as outlined in Scheme 2. Whereas enantioselective synthesis of 2-substituted formylcymantrenes 5a–c, precursors to 1a–c, were reported [33-35], racemic 5a–c were prepared in the same ways starting with rac-3. Vinylcymantrene substrates rac-1a–c were obtained in 68–79% yields by the Wittig methylenation of rac-5a–c. The moderate yields could be attributed to the volatility of rac-1a–c.
![[1860-5397-22-42-i2]](/bjoc/content/inline/1860-5397-22-42-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of racemic planar-chiral vinylcymantrene substrates rac-1a–c.
Scheme 2: Preparation of racemic planar-chiral vinylcymantrene substrates rac-1a–c.
Molybdenum-catalyzed asymmetric metathesis dimerization (AMD)/kinetic resolution (KR) of racemic planar-chiral vinylcymantrene derivatives rac-1a–c
The vinylcymantrene substrates, rac-1, prepared as above are planar-chiral due to the presence of an unsymmetrically substituted η5-(1-R-2-vinylcyclopentadienyl ligand (R = Br, Me, or I). The racemic substrates were examined in the AMD/KR studies, and the results are summarized in Table 1.
Table 1: Molybdenum-catalyzed asymmetric metathesis dimerization/kinetic resolution of racemic vinylcymantrenes 1a–c.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-22-42-i3.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | Subst. | Chiral ligand | Conditions | Conversion | chiral-2/ meso-2b | % eed | krelc | ||
| Exptl.b | Calcd.c | 1 | chiral-2 | ||||||
| 1 | rac-1a | (R)-L1 | 60 °C, 24 h | 25% | 26% | 97:3 | 34 | 95 | 86 |
| 2 | rac-1a | (R)-L2 | 60 °C, 24 h | 3% | –e | –e | 0.7 | 66 | 4.9 |
| 3 | rac-1a | (R)-L3 | 60 °C, 24 h | 51% | 34% | 85:15 | 48 | 95 | 107 |
| 4 | rac-1a | (R)-L1 | 10 °C, 48 h | 37% | 31% | 96:4 | 45 | 99 | 754 |
| 5 | rac-1a | (R)-L1 | 10 °C, 72 h | 18% | 21% | 98:2 | 26 | 97 | 120 |
| 6 | rac-1a | (R)-L3 | 10 °C, 48 h | 46% | 42% | 94:6 | 65 | 92 | 120 |
| 7 | rac-1a | (R)-L3 | 10 °C, 72 h | 23% | 26% | 96:4 | 33 | 92 | 47 |
| 8 | rac-1b | (R)-L1 | 10 °C, 48 h | 40% | 32% | >99.5:<0.5 | 46 | 99 | 489 |
| 9 | rac-1b | (R)-L3 | 10 °C, 48 h | 58% | 59% | 73:27 | 74 | 88 | 21 |
| 10 | rac-1c | (R)-L1 | 10 °C, 48 h | 21% | 29% | >99.5/<0.5 | 40 | >99.5 | 1550 |
| 11 | rac-1c | (R)-L3 | 10 °C, 48 h | 29% | 32% | >99.5:<0.5 | 47 | 98 | 286 |
| 12 | rac-1c | (R)-L3 | 60 °C, 24 h | 35% | 29% | >99.5:<0.5 | 41 | 99 | 340 |
aThe reaction was carried out with rac-1 (0.20 mmol) in benzene (3.0 mL) for 24 h in the presence of a molybdenum catalyst generated in situ (10 mol %, except 5 mol % in entries 5 and 7); bdetermined by the 1H NMR analysis of the crude reaction mixture; ccalculated from the % ee values of recovered 1 and chiral-2 based on a second-order equation (refs. [37] and [38]; see Supporting Information File 1 for details); ddetermined by chiral HPLC (see Supporting Information File 1 for details); enot determined.
At the outset, the optimization of the reaction conditions, including a proper choice of chiral molybdenum-alkylidene precatalysts, was examined using rac-1a as a prototypical substrate. The AMD/KR reactions were conducted in benzene at the indicated temperature in the presence of an appropriate chiral Mo-precatalyst (10 mol %), which was generated in situ from the Mo-precursor, (pyrrolyl)2Mo(=CHCMe2Ph)(=N-C6H3-2,6-iPr2) [36], and an axially chiral biphenol ligand. The chiral Mo/(R)-L1 precatalyst [21] facilitated the metathesis dimerization of (R)-1a preferentially, and the most of antipodal (S)-1a was recovered intact. In general, vinylcymantrenes are much less reactive than analogous vinylferrocenes in the molybdenum-catalyzed AMD [30], and the conversion tends to be lower. Only 25% conversion was attained in the reaction at 60 °C for 24 h. The crude reaction mixture was analyzed by 1H NMR measurement, which revealed the presence of the two diastereomers in the dimerized product with the molar ratio of chiral-2a/meso-2a = 97:3. The enantiomeric purity of recovered (S)-1a was determined to be 34% ee, while that of (R,R)-2a was found to be 95% ee (Table 1, entry 1). The krel value for the reaction was calculated to be 86 based on a second-order equation [30,31,37,38]. The Mo/(R)-L2 precatalyst [22] was much less active than Mo/(R)-L1 and less than 3% conversion was attained under the otherwise identical conditions (Table 1, entry 2). The Mo/(R)-L3 precatalyst [23] showed the highest catalytic activity among examined. Although the enantioselectivity was excellent (krel = 107), the diastereoselectivity was poor (Table 1, entry 3). At 10 °C using Mo/(R)-L1, enantioselectivity was greatly improved in the AMD/KR of rac-1a giving (R,R)-2a of 99% ee and (S)-1a of 45% ee with 37% conversion (krel = 754; Table 1, entry 4). The lower catalyst loading (5 mol %) led to an unsatisfactory conversion (18%) probably due to the decomposition of the molybdenum catalyst prior to the completion of the reaction (Table 1, entry 5). On the other hand, the effects of lowering the temperature were minimal in the reactions using Mo/(R)-L3 (Table 1, entries 6 and 7).
The optimized conditions as in entries 4 and 6 were applied to the AMD/KR reactions of rac-1b and 1c as well. The AMD/KR of rac-1b using Mo/(R)-L1 proceeded with excellent diastereo-/enantioselectivities (chiral-2b/meso-2b = >99.5:<0.5; krel = 489), and (R)-1b and (S,S)-2b were obtained in 46% ee and 99% ee, respectively (Table 1, entry 8). On the other hand, the results using Mo/(R)-L3 were poor with chiral-2b/meso-2b = 73:27 and krel = 21 (Table 1, entry 9). The AMD reaction of rac-1c was sluggish. The conversion at 10 °C in 48 h was only 21% using Mo/(R)-L1 (krel = 1.5 × 103; chiral-2c/meso-2c = >99.5:<0.5; Table 1, entry 10) and 29% using Mo/(R)-L3 (krel = 286; chiral-2c/meso-2c = >99.5:<0.5; Table 1, entry 11) despite the excellent enantio- and diastereoselectivities. The conversion was improved to 35% at 60 °C using Mo/(R)-L3, and the excellent enantio- and diastereoselectivities were retained (krel = 340; chiral-2c/meso-2c = >99.5:<0.5; Table 1, entry 12). All the AMD products were obtained in E-configurations exclusively.
Determination of the absolute configuration of (–)-2b
Levorotatory AMD/KR product (–)-2b, which was obtained as in entry 8, Table 1 using Mo/(R)-L1 precatalyst, was recrystallized by slow diffusion of pentane into the concentrated diethyl ether solution. Crystals of (–)-2b were grown as light-yellow blocks. The X-ray crystallography revealed that the unit cell contains two independent molecules, having slightly different conformations, and the structure of one of the two crystallographically independent molecules is shown in Figure 1 with the selected bond lengths and angles (see Supporting Information File 1 and Supporting Information File 2 for details).
![[1860-5397-22-42-1]](/bjoc/content/figures/1860-5397-22-42-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP drawing of the X-ray structure of (S,S)-(–)-2b with atom numbering (thermal ellipsoids set at the 30% probability level). Selected bond lengths (Å) and angles (deg): C2–C13 = 1.460(3), C8–C14 = 1.461(3), C13–C140 = 1.340(3), Mn1-least-squares planeC1–C5 = 1.771 (7), Mn2-least-squares planeC7–C11 = 1.770(7); C2–C13–C14 = 123.6(2), C8–C14–C13 = 125.7(2), dihedral angle between least-squares planeC1–C5 and least-squares planeC7–C11 = 17.40(8).
Figure 1: ORTEP drawing of the X-ray structure of (S,S)-(–)-2b with atom numbering (thermal ellipsoids set at...
The two cyclopentadienides and the olefinic moiety are nearly coplanar with a C2–C13–C14–C8 torsion angle of 175.4(2)°. The Flack parameter for this structure was determined to be –0.010(4), and the absolute configuration of (–)-2b was unambiguously determined to be (S,S) (see Supporting Information File 1 and Supporting Information File 2 for details).
Comparison of vinylcymantrenes, vinylferrocenes, and vinylphosphaferrocenes in molybdenum-catalyzed AMD/KR
Figure 2 shows the structures of the less-reactive enantiomers in the three representative substrates (vinylcymantrene, vinylferrocene [30], and vinylphosphaferrocene [31]) in the AMD/KR reactions using Mo/(R)-L1. The views of the molecules from the view point opposite to the central metal cations (Mn(I) or Fe(II)) are illustrated at the bottom of Figure 2. All the three compounds possess the similar structural features: (1) presence of a substituent (Br or Me) at the position adjacent to the vinyl group in the clockwise direction, (2) absence of a substituent other than hydrogen at the position adjacent to the vinyl group in the counterclockwise direction (CH or P). The substituent adjacent to the vinyl group (marked in red in Figure 2) likely inhibits the effective interaction of the substrate with the chiral catalyst, resulting in highly enantioselective kinetic resolution.
![[1860-5397-22-42-2]](/bjoc/content/figures/1860-5397-22-42-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of less-reactive enantiomers in three representative planar-chiral vinylmetallocene substrates in Mo/(R)-L1-catalyzed AMD/KR.
Figure 2: Structures of less-reactive enantiomers in three representative planar-chiral vinylmetallocene subs...
Cymantrene is far less electron-poor than ferrocene due to the presence of the three carbonyl ligands, which are strong π-acids, on the manganese(I). Indeed, the Friedel–Crafts acetylation of cymantrene, a typical electrophilic aromatic substitution reaction, is much slower than that of ferrocene. Consequently, vinylcymantrenes are electron-deficient olefins and less reactive in olefin metathesis. For this reason, the present AMD/KR reactions of rac-1a–c require a relatively high catalyst loading, resulting in lower conversions.
Conclusion
In summary, we have developed a protocol for the kinetic resolution (KR) of racemic planar-chiral 1-R-2-vinylcymantrenes (rac-1; R = Br, Me, I) by the molybdenum-catalyzed asymmetric metathesis dimerization (AMD). The AMD/KR reactions of rac-1 proceeded with near-perfect diastereoselectivity, of which enantioselectivity was also excellent with the krel values of up to 1.5 × 103. Because of the outstanding enantioselectivity, dimerized products chiral-2 were obtained in very high enantiomeric purity of up to >99% ee. Vinylcymantrenes are electron-deficient olefins, and they are poorer substrates in olefin metathesis. For this reason, the AMD/KR reactions of rac-1a–c require a relatively high catalyst loading and the conversions tend to be lower.
This study, together with our previous reports [29], reveals that the molybdenum-catalyzed asymmetric metathesis reactions are powerful tools to control planar chirality in various transition-metal complexes.
Supporting Information
| Supporting Information File 1: Experimental procedures, NMR spectra (1H and 13C) for all the new compounds, and chiral HPLC chromatograms. | ||
| Format: PDF | Size: 3.5 MB | Download |
| Supporting Information File 2: Crystallographic information file for compound 2b. | ||
| Format: CIF | Size: 1.1 MB | Download |
Funding
This work was supported by Grants-in-Aid for Scientific Research (23H01974 and 25K22274 for Y.O., 23K13763 and 25K18050 for H.I., and 21H01940 for M.O.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT), CREST grant (JPMJCR21B1 for Y.O.) from JST, the Yazaki Memorial Foundation and Toshiaki Ogasawara Memorial Foundation (for Y.O.). This work was also supported by the International Collaborative Research Program of the Institute for Chemical Research, Kyoto University(for H.I., Y.O., and M.O.)
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Schrock, R. R.; Murdzek, J. S.; Bazan, G. C.; Robbins, J.; DiMare, M.; O'Regan, M. J. Am. Chem. Soc. 1990, 112, 3875–3886. doi:10.1021/ja00166a023
Return to citation in text: [1] -
Schrock, R. R. The Discovery and Development of High Oxidation State Mo and W Imido Alkylidene Complexes for Alkene Metathesis. In Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 8–32. doi:10.1002/9783527619481.ch3
Return to citation in text: [1] -
Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d
Return to citation in text: [1] -
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q
Return to citation in text: [1] -
Nguyen, S. T.; Trnka, T. M. The Discovery and Development of Well-Defined, Ruthenium-Based Olefin Metathesis Catalysts. In Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 61–85. doi:10.1002/9783527619481.ch6
Return to citation in text: [1] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576
Return to citation in text: [1] -
Klare, H. F. T.; Oestreich, M. Angew. Chem., Int. Ed. 2009, 48, 2085–2089. doi:10.1002/anie.200806254
Return to citation in text: [1] -
Kress, S.; Blechert, S. Chem. Soc. Rev. 2012, 41, 4389–4408. doi:10.1039/c2cs15348c
Return to citation in text: [1] -
Hoveyda, A. H. J. Org. Chem. 2014, 79, 4763–4792. doi:10.1021/jo500467z
Return to citation in text: [1] -
Ogasawara, M.; Nagano, T.; Hayashi, T. J. Am. Chem. Soc. 2002, 124, 9068–9069. doi:10.1021/ja026401r
(correction: J. Am. Chem. Soc. 2002, 124, 12626. doi:10.1021/ja028166i)
Return to citation in text: [1] -
Ogasawara, M.; Nagano, T.; Hayashi, T. Organometallics 2003, 22, 1174–1176. doi:10.1021/om021058k
Return to citation in text: [1] -
Locke, A. J.; Jones, C.; Richards, C. J. J. Organomet. Chem. 2001, 637–639, 669–676. doi:10.1016/s0022-328x(01)00980-9
Return to citation in text: [1] -
Hüerländer, D.; Kleigrewe, N.; Kehr, G.; Erker, G.; Fröhlich, R. Eur. J. Inorg. Chem. 2002, 2633–2642. doi:10.1002/1099-0682(200210)2002:10<2633::aid-ejic2633>3.0.co;2-4
Return to citation in text: [1] -
Ogasawara, M.; Watanabe, S.; Fan, L.; Nakajima, K.; Takahashi, T. Organometallics 2006, 25, 5201–5203. doi:10.1021/om0608298
Return to citation in text: [1] -
Ogasawara, M.; Wu, W.-Y.; Arae, S.; Watanabe, S.; Morita, T.; Takahashi, T.; Kamikawa, K. Angew. Chem., Int. Ed. 2012, 51, 2951–2955. doi:10.1002/anie.201108292
Return to citation in text: [1] -
Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Chem. – Eur. J. 2013, 19, 4151–4154. doi:10.1002/chem.201300116
Return to citation in text: [1] -
Ogasawara, M.; Watanabe, S.; Nakajima, K.; Takahashi, T. J. Am. Chem. Soc. 2010, 132, 2136–2137. doi:10.1021/ja910348z
Return to citation in text: [1] -
Kamikawa, K.; Arae, S.; Wu, W.-Y.; Nakamura, C.; Takahashi, T.; Ogasawara, M. Chem. – Eur. J. 2015, 21, 4954–4957. doi:10.1002/chem.201500226
Return to citation in text: [1] -
Ogasawara, M.; Tseng, Y.-Y.; Uryu, M.; Ohya, N.; Chang, N.; Ishimoto, H.; Arae, S.; Takahashi, T.; Kamikawa, K. Organometallics 2017, 36, 4061–4069. doi:10.1021/acs.organomet.7b00704
Return to citation in text: [1] [2] -
Aeilts, S. L.; Cefalo, D. R.; Bonitatebus, P. J., Jr.; Houser, J. H.; Hoveyda, A. H.; Schrock, R. R. Angew. Chem., Int. Ed. 2001, 40, 1452–1456. doi:10.1002/1521-3773(20010417)40:8<1452::aid-anie1452>3.0.co;2-g
Return to citation in text: [1] -
Schrock, R. R.; Jamieson, J. Y.; Dolman, S. J.; Miller, S. A.; Bonitatebus,, P. J.; Hoveyda, A. H. Organometallics 2002, 21, 409–417. doi:10.1021/om0107441
Return to citation in text: [1] [2] -
Alexander, J. B.; La, D. S.; Cefalo, D. R.; Hoveyda, A. H.; Schrock, R. R. J. Am. Chem. Soc. 1998, 120, 4041–4042. doi:10.1021/ja974353i
Return to citation in text: [1] [2] -
Singh, R.; Czekelius, C.; Schrock, R. R.; Müller, P.; Hoveyda, A. H. Organometallics 2007, 26, 2528–2539. doi:10.1021/om061134+
Return to citation in text: [1] [2] -
Halterman, R. L. Chem. Rev. 1992, 92, 965–994. doi:10.1021/cr00013a011
Return to citation in text: [1] -
Togni, A. Angew. Chem., Int. Ed. Engl. 1996, 35, 1475–1477. doi:10.1002/anie.199614751
Return to citation in text: [1] -
Schaarschmidt, D.; Lang, H. Organometallics 2013, 32, 5668–5704. doi:10.1021/om400564x
Return to citation in text: [1] -
Arae, S.; Ogasawara, M. Tetrahedron Lett. 2015, 56, 1751–1761. doi:10.1016/j.tetlet.2015.01.130
Return to citation in text: [1] -
Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2017, 50, 351–365. doi:10.1021/acs.accounts.6b00573
Return to citation in text: [1] -
Ogasawara, M. Chem. Rec. 2021, 21, 3509–3519. doi:10.1002/tcr.202100102
Return to citation in text: [1] [2] -
Nishimoto, K.; Taue, H.; Ohji, T.; Funakoshi, S.; Ohki, Y.; Ogasawara, M. Org. Lett. 2022, 24, 7355–7360. doi:10.1021/acs.orglett.2c02888
Return to citation in text: [1] [2] [3] [4] [5] -
Masaoka, K.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. Organometallics 2023, 42, 1629–1638. doi:10.1021/acs.organomet.3c00185
Return to citation in text: [1] [2] [3] [4] -
Ogasawara, M.; Tseng, Y.-Y.; Liu, Q.; Chang, N.; Yang, X.; Takahashi, T.; Kamikawa, K. Organometallics 2017, 36, 1430–1435. doi:10.1021/acs.organomet.7b00125
Return to citation in text: [1] -
Kamikawa, K.; Tseng, Y.-Y.; Jian, J.-H.; Takahashi, T.; Ogasawara, M. J. Am. Chem. Soc. 2017, 139, 1545–1553. doi:10.1021/jacs.6b11243
Return to citation in text: [1] -
Loim, N. M.; Kondratenko, M. A.; Sokolov, V. I. J. Org. Chem. 1994, 59, 7485–7487. doi:10.1021/jo00103a049
Return to citation in text: [1] -
Ferber, B.; Top, S.; Jaouen, G. J. Organomet. Chem. 2004, 689, 4872–4876. doi:10.1016/j.jorganchem.2004.09.057
Return to citation in text: [1] -
Hock, A. S.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2006, 128, 16373–16375. doi:10.1021/ja0665904
Return to citation in text: [1] -
Kagan, H. B.; Fiaud, J. C. Kinetic Resolution. In Topics in Stereochemistry; Eliel, E. L.; Wilen, S. H., Eds.; John Wiley & Sons: New York, NY, USA, 1988; Vol. 18, pp 249–330. doi:10.1002/9780470147276.ch4
Return to citation in text: [1] [2] -
Luukas, T. O.; Girard, C.; Fenwick, D. R.; Kagan, H. B. J. Am. Chem. Soc. 1999, 121, 9299–9306. doi:10.1021/ja990793t
Return to citation in text: [1] [2]
| 30. | Nishimoto, K.; Taue, H.; Ohji, T.; Funakoshi, S.; Ohki, Y.; Ogasawara, M. Org. Lett. 2022, 24, 7355–7360. doi:10.1021/acs.orglett.2c02888 |
| 31. | Masaoka, K.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. Organometallics 2023, 42, 1629–1638. doi:10.1021/acs.organomet.3c00185 |
| 37. | Kagan, H. B.; Fiaud, J. C. Kinetic Resolution. In Topics in Stereochemistry; Eliel, E. L.; Wilen, S. H., Eds.; John Wiley & Sons: New York, NY, USA, 1988; Vol. 18, pp 249–330. doi:10.1002/9780470147276.ch4 |
| 38. | Luukas, T. O.; Girard, C.; Fenwick, D. R.; Kagan, H. B. J. Am. Chem. Soc. 1999, 121, 9299–9306. doi:10.1021/ja990793t |
| 21. | Schrock, R. R.; Jamieson, J. Y.; Dolman, S. J.; Miller, S. A.; Bonitatebus,, P. J.; Hoveyda, A. H. Organometallics 2002, 21, 409–417. doi:10.1021/om0107441 |
| 30. | Nishimoto, K.; Taue, H.; Ohji, T.; Funakoshi, S.; Ohki, Y.; Ogasawara, M. Org. Lett. 2022, 24, 7355–7360. doi:10.1021/acs.orglett.2c02888 |
| 1. | Schrock, R. R.; Murdzek, J. S.; Bazan, G. C.; Robbins, J.; DiMare, M.; O'Regan, M. J. Am. Chem. Soc. 1990, 112, 3875–3886. doi:10.1021/ja00166a023 |
| 2. | Schrock, R. R. The Discovery and Development of High Oxidation State Mo and W Imido Alkylidene Complexes for Alkene Metathesis. In Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 8–32. doi:10.1002/9783527619481.ch3 |
| 14. | Ogasawara, M.; Watanabe, S.; Fan, L.; Nakajima, K.; Takahashi, T. Organometallics 2006, 25, 5201–5203. doi:10.1021/om0608298 |
| 15. | Ogasawara, M.; Wu, W.-Y.; Arae, S.; Watanabe, S.; Morita, T.; Takahashi, T.; Kamikawa, K. Angew. Chem., Int. Ed. 2012, 51, 2951–2955. doi:10.1002/anie.201108292 |
| 16. | Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Chem. – Eur. J. 2013, 19, 4151–4154. doi:10.1002/chem.201300116 |
| 38. | Luukas, T. O.; Girard, C.; Fenwick, D. R.; Kagan, H. B. J. Am. Chem. Soc. 1999, 121, 9299–9306. doi:10.1021/ja990793t |
| 10. |
Ogasawara, M.; Nagano, T.; Hayashi, T. J. Am. Chem. Soc. 2002, 124, 9068–9069. doi:10.1021/ja026401r
(correction: J. Am. Chem. Soc. 2002, 124, 12626. doi:10.1021/ja028166i) |
| 11. | Ogasawara, M.; Nagano, T.; Hayashi, T. Organometallics 2003, 22, 1174–1176. doi:10.1021/om021058k |
| 12. | Locke, A. J.; Jones, C.; Richards, C. J. J. Organomet. Chem. 2001, 637–639, 669–676. doi:10.1016/s0022-328x(01)00980-9 |
| 13. | Hüerländer, D.; Kleigrewe, N.; Kehr, G.; Erker, G.; Fröhlich, R. Eur. J. Inorg. Chem. 2002, 2633–2642. doi:10.1002/1099-0682(200210)2002:10<2633::aid-ejic2633>3.0.co;2-4 |
| 36. | Hock, A. S.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2006, 128, 16373–16375. doi:10.1021/ja0665904 |
| 6. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576 |
| 7. | Klare, H. F. T.; Oestreich, M. Angew. Chem., Int. Ed. 2009, 48, 2085–2089. doi:10.1002/anie.200806254 |
| 8. | Kress, S.; Blechert, S. Chem. Soc. Rev. 2012, 41, 4389–4408. doi:10.1039/c2cs15348c |
| 9. | Hoveyda, A. H. J. Org. Chem. 2014, 79, 4763–4792. doi:10.1021/jo500467z |
| 33. | Kamikawa, K.; Tseng, Y.-Y.; Jian, J.-H.; Takahashi, T.; Ogasawara, M. J. Am. Chem. Soc. 2017, 139, 1545–1553. doi:10.1021/jacs.6b11243 |
| 34. | Loim, N. M.; Kondratenko, M. A.; Sokolov, V. I. J. Org. Chem. 1994, 59, 7485–7487. doi:10.1021/jo00103a049 |
| 35. | Ferber, B.; Top, S.; Jaouen, G. J. Organomet. Chem. 2004, 689, 4872–4876. doi:10.1016/j.jorganchem.2004.09.057 |
| 3. | Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d |
| 4. | Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q |
| 5. | Nguyen, S. T.; Trnka, T. M. The Discovery and Development of Well-Defined, Ruthenium-Based Olefin Metathesis Catalysts. In Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 61–85. doi:10.1002/9783527619481.ch6 |
| 37. | Kagan, H. B.; Fiaud, J. C. Kinetic Resolution. In Topics in Stereochemistry; Eliel, E. L.; Wilen, S. H., Eds.; John Wiley & Sons: New York, NY, USA, 1988; Vol. 18, pp 249–330. doi:10.1002/9780470147276.ch4 |
| 30. | Nishimoto, K.; Taue, H.; Ohji, T.; Funakoshi, S.; Ohki, Y.; Ogasawara, M. Org. Lett. 2022, 24, 7355–7360. doi:10.1021/acs.orglett.2c02888 |
| 31. | Masaoka, K.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. Organometallics 2023, 42, 1629–1638. doi:10.1021/acs.organomet.3c00185 |
| 31. | Masaoka, K.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. Organometallics 2023, 42, 1629–1638. doi:10.1021/acs.organomet.3c00185 |
| 30. | Nishimoto, K.; Taue, H.; Ohji, T.; Funakoshi, S.; Ohki, Y.; Ogasawara, M. Org. Lett. 2022, 24, 7355–7360. doi:10.1021/acs.orglett.2c02888 |
| 24. | Halterman, R. L. Chem. Rev. 1992, 92, 965–994. doi:10.1021/cr00013a011 |
| 25. | Togni, A. Angew. Chem., Int. Ed. Engl. 1996, 35, 1475–1477. doi:10.1002/anie.199614751 |
| 26. | Schaarschmidt, D.; Lang, H. Organometallics 2013, 32, 5668–5704. doi:10.1021/om400564x |
| 27. | Arae, S.; Ogasawara, M. Tetrahedron Lett. 2015, 56, 1751–1761. doi:10.1016/j.tetlet.2015.01.130 |
| 28. | Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2017, 50, 351–365. doi:10.1021/acs.accounts.6b00573 |
| 29. | Ogasawara, M. Chem. Rec. 2021, 21, 3509–3519. doi:10.1002/tcr.202100102 |
| 19. | Ogasawara, M.; Tseng, Y.-Y.; Uryu, M.; Ohya, N.; Chang, N.; Ishimoto, H.; Arae, S.; Takahashi, T.; Kamikawa, K. Organometallics 2017, 36, 4061–4069. doi:10.1021/acs.organomet.7b00704 |
| 32. | Ogasawara, M.; Tseng, Y.-Y.; Liu, Q.; Chang, N.; Yang, X.; Takahashi, T.; Kamikawa, K. Organometallics 2017, 36, 1430–1435. doi:10.1021/acs.organomet.7b00125 |
| 31. | Masaoka, K.; Taue, H.; Wakioka, M.; Ohki, Y.; Ogasawara, M. Organometallics 2023, 42, 1629–1638. doi:10.1021/acs.organomet.3c00185 |
| 20. | Aeilts, S. L.; Cefalo, D. R.; Bonitatebus, P. J., Jr.; Houser, J. H.; Hoveyda, A. H.; Schrock, R. R. Angew. Chem., Int. Ed. 2001, 40, 1452–1456. doi:10.1002/1521-3773(20010417)40:8<1452::aid-anie1452>3.0.co;2-g |
| 21. | Schrock, R. R.; Jamieson, J. Y.; Dolman, S. J.; Miller, S. A.; Bonitatebus,, P. J.; Hoveyda, A. H. Organometallics 2002, 21, 409–417. doi:10.1021/om0107441 |
| 22. | Alexander, J. B.; La, D. S.; Cefalo, D. R.; Hoveyda, A. H.; Schrock, R. R. J. Am. Chem. Soc. 1998, 120, 4041–4042. doi:10.1021/ja974353i |
| 23. | Singh, R.; Czekelius, C.; Schrock, R. R.; Müller, P.; Hoveyda, A. H. Organometallics 2007, 26, 2528–2539. doi:10.1021/om061134+ |
| 22. | Alexander, J. B.; La, D. S.; Cefalo, D. R.; Hoveyda, A. H.; Schrock, R. R. J. Am. Chem. Soc. 1998, 120, 4041–4042. doi:10.1021/ja974353i |
| 17. | Ogasawara, M.; Watanabe, S.; Nakajima, K.; Takahashi, T. J. Am. Chem. Soc. 2010, 132, 2136–2137. doi:10.1021/ja910348z |
| 18. | Kamikawa, K.; Arae, S.; Wu, W.-Y.; Nakamura, C.; Takahashi, T.; Ogasawara, M. Chem. – Eur. J. 2015, 21, 4954–4957. doi:10.1002/chem.201500226 |
| 19. | Ogasawara, M.; Tseng, Y.-Y.; Uryu, M.; Ohya, N.; Chang, N.; Ishimoto, H.; Arae, S.; Takahashi, T.; Kamikawa, K. Organometallics 2017, 36, 4061–4069. doi:10.1021/acs.organomet.7b00704 |
| 30. | Nishimoto, K.; Taue, H.; Ohji, T.; Funakoshi, S.; Ohki, Y.; Ogasawara, M. Org. Lett. 2022, 24, 7355–7360. doi:10.1021/acs.orglett.2c02888 |
| 23. | Singh, R.; Czekelius, C.; Schrock, R. R.; Müller, P.; Hoveyda, A. H. Organometallics 2007, 26, 2528–2539. doi:10.1021/om061134+ |
© 2026 Imazu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.