Abstract
An organosilicon cation stabilized by intramolecular pyridyl coordination was effectively generated and accumulated by oxidative Si-Si bond dissociation of the corresponding disilane using low temperature electrolysis, and was characterized by NMR and CSI-MS.

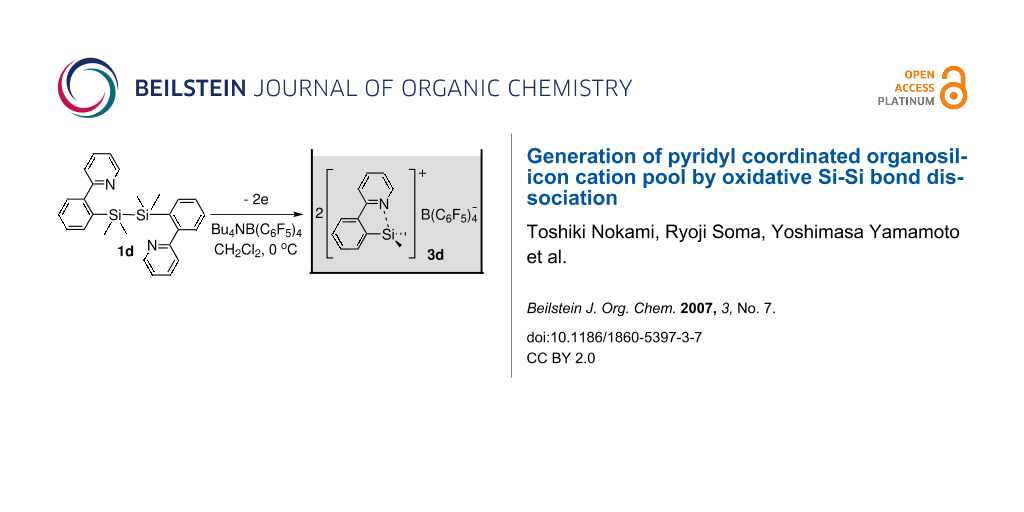
Graphical Abstract
Findings
We have recently developed the "cation pool" method, which involves the irreversible oxidative generation and accumulation of highly reactive cations in the absence of nucleophiles [1-5]. Heteroatom-stabilized carbocations, such as N-acyliminium ion pools and alkoxycarbenium ion pools have been generated based on oxidative C-H, C-Si, and C-S bond dissociation. Very recently, the oxidative C-C bond dissociation has been found to be effective for generation of a pool of a carbocation having a stabilizing group as shown in Scheme 1[6].
![[1860-5397-3-7-i1]](/bjoc/content/inline/1860-5397-3-7-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Electrochemical generation of carbocations by oxidative C-C bond dissociation.
Scheme 1: Electrochemical generation of carbocations by oxidative C-C bond dissociation.
We have been interested in generation and accumulation of cations of other elements such as silicon using the "cation pool" method. Organosilicon cations are known to be extremely unstable and difficult to accumulate in solution [7-11]. Organosilicon cations having appropriate donor ligands are, however, reasonably stable to accumulate in solution and many examples of such donor-stabilized organosilicon cations have been reported in the literature [12-18]. Herein, we report the generation and accumulation of a donor-stabilized organosilicon cation by the electrochemical oxidative Si-Si bond dissociation (Scheme 2) [19-21].
![[1860-5397-3-7-i2]](/bjoc/content/inline/1860-5397-3-7-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electrochemical generation and accumulation of organosilicon cation by oxidative Si-Si bond dissociation.
Scheme 2: Electrochemical generation and accumulation of organosilicon cation by oxidative Si-Si bond dissoci...
Symmetrical disilanes having coordinating groups on both silicon atoms were used as starting materials for electrochemical generation and accumulation of organosilicon cations, because oxidative dissociation of the Si-Si bond leads to the formation of two equivalents of organosilicon cations and no other product is formed.
In our earlier study, it was found that the introduction of a coordinating group such as a pyridyl group decreased the oxidation potential of tetraalkylstannanes, although there is no indication of the coordination in the neutral molecule. Dynamic intramolecular coordination to tin seems to facilitate electron transfer [22]. The coordination also stabilizes the thus-generated radical cation and weakens the C-Sn bond. A similar effect of intramolecular coordination was observed in the case of silicon [23]. Another important point is that pyridyl group is rather inactive toward the anodic oxidation. Thus, we chose to use a pyridyl group as a donor ligand.
First, we prepared disilanes having pyridyl groups in appropriate positions and measured their oxidation potentials [24,25]. The oxidation potential of 2-pyridylethyl substituted disilane 1b was slightly less positive than hexamethyldisilane 1a. On the other hand, the oxidation potential of 2-pyridylphenyl substituted disilane 1d was much less positive than the corresponding disilane 1c having phenyl groups (Figure 1).
![[1860-5397-3-7-1]](/bjoc/content/figures/1860-5397-3-7-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Oxidation potentials (Ed; decomposition potential) of disilanes determined by rotating disk electrode voltammetry (RDE) in Bu4NClO4/CH3CN.
Figure 1: Oxidation potentials (Ed; decomposition potential) of disilanes determined by rotating disk electro...
29Si NMR chemical shifts of 1b and 1d were similar to those of 1a and 1c, indicating that no coordination of the pyridyl groups on silicon existed in the neutral molecules (Supporting Information File 1). Therefore, the significant effect of the 2-pyridyl group on the oxidation potential may be ascribed to effective intramolecular coordination to stabilize the radical cation intermediate. The conformationally less flexible 2-pyridylphenyl group seems to be more effective than the 2-pyridylethyl group.
The intramolecular coordination in the radical cation is supported by the DFT calculations as shown in Figure 2. It is also important to note that such coordination elongates the Si-Si bond and facilitates its dissociation.
![[1860-5397-3-7-2]](/bjoc/content/figures/1860-5397-3-7-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Optimized structures of radical cations of 1,2-diphenyldisilane and 1,2-bis [o-(2-pyridyl)- phenyl]disilane obtained by DFT calculation (B3LYP/LANL2DZ).
Figure 2: Optimized structures of radical cations of 1,2-diphenyldisilane and 1,2-bis [o-(2-pyridyl)- phenyl]...
Preparative electrochemical oxidation of 1d was carried out to generate and accumulate the corresponding organosilicon cation 3d (Scheme 3). Nature of the counter anion was very important. When 1d was oxidized in the presence of Bu4NBF4, which is a common supporting electrolyte for the "cation pool" method, fluoride was introduced on the silicon atom. Eventually, Bu4NB(C6F5)4 was found to be an appropriate supporting electrolyte to generate and accumulate the organosilicon cation 3d.
![[1860-5397-3-7-i3]](/bjoc/content/inline/1860-5397-3-7-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Electrochemical generation of organosilicon cation 3d.
Scheme 3: Electrochemical generation of organosilicon cation 3d.
The 1H NMR spectrum of the solution obtained by the electrochemical oxidation of 1d in CH2Cl2 (containing 10% CD2Cl2) using Bu4NB(C6F5)4 at 0°C showed complete conversion of disilane 1d to one species, i.e. organosilicon cation 3d. The Si-CH3 groups in 3d exhibited a signal at 0.87 ppm, whereas those in 1d were observed at -0.05 ppm. Significant low field shift of the protons on the pyridyl ring was also observed. 3d exhibited a 29Si signal at 37.7 ppm [26-28]. These results strongly suggest the generation of an electron deficient silicon species. Therefore, it is reasonable to consider that the organosilicon cation stabilized by the pyridyl coordination was generated.
The formation of organosilicon cation 3d was also confirmed by CSI-MS (cold-spray ionization mass spectroscopy) (spray temperature; 0°C) [29]. The parent peak was observed at M/Z = 212.08963 (Calcd: 212.08955) as shown in Figure 3. A complex of 3d with HF was also observed, although the mechanism of its formation is not clear at present.
![[1860-5397-3-7-3]](/bjoc/content/figures/1860-5397-3-7-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CSI-MS of organosilicon cation 3d at 0°C.
Figure 3: CSI-MS of organosilicon cation 3d at 0°C.
The mechanism shown in Figure 4 seems to be reasonable. The initial one-electron oxidation of disilane 1d gives radical cation 2d. The pyridyl coordination in the radical cation facilitates the electron transfer. In the next step, the dissociation of the Si-Si bond in radical cation 2d takes place to give organosilicon cation 3d and silyl radical 4d. DFT calculations indicated that the pyridyl group coordination to silicon takes place both in cation and radical. Radical 4d seems to be easily oxidized on the surface of the electrode to give cation 3d. Therefore, two moles of 3d should be formed from one mole of 1d by net two-electron oxidation.
![[1860-5397-3-7-4]](/bjoc/content/figures/1860-5397-3-7-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Optimized structures of organosilicon cation 3d and silyl radical 4d by DFT calculations (B3LYP/LANL2DZ).
Figure 4: Optimized structures of organosilicon cation 3d and silyl radical 4d by DFT calculations (B3LYP/LAN...
The organosilicon cation 3d can be trapped by p-tolylmagnesium bromide as a nucleophile and the corresponding product 5d was obtained in 90% yield based on disilane 1d (Scheme 4) [30]. The observation indicates that two moles of the cation is formed from one mole of the disilane with the consumption of two moles of electrons, being consistent with the mechanism shown in Figure 4.
![[1860-5397-3-7-i4]](/bjoc/content/inline/1860-5397-3-7-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of organosilicon cation 3d with p-tolylmagnesium bromide.
Scheme 4: Reaction of organosilicon cation 3d with p-tolylmagnesium bromide.
Effective formation of 5d indicates that organosilicon cation 3d acted as a silicon centered cation. The carbon nucleophile attacked the silicon atom selectively, although a positive charge should also be delocalized on the nitrogen atom.
The present observations speak well for possibilities of the "cation pool" method in organosilicon chemistry. Donor-stabilized organosilicon cations can be effectively generated and accumulated at 0°C by the electrochemical oxidative Si-Si bond dissociation. It is also noteworthy that the presence of a donor ligand on the silicon atom facilitates the oxidation. Further work aimed at generating other organosilicon cations and exploring their stability and reactivity is currently in progress.
Supporting Information
| Supporting Information File 1: Supporting information. Experimental procedures, spectrum data of new compounds, details of DFT calculation, and 1H/13C NMR spectra. | ||
| Format: PDF | Size: 621.1 KB | Download |
References
-
Yoshida, J.; Suga, S. Chem.–Eur. J. 2002, 8, 2650–2658. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S
Return to citation in text: [1] -
Suga, S.; Watanabe, M.; Yoshida, J. J. Am. Chem. Soc. 2002, 124, 14824–14825. doi:10.1021/ja028663z
Return to citation in text: [1] -
Suga, S.; Suzuki, S.; Maruyama, T.; Yoshida, J. Bull. Chem. Soc. Jpn. 2004, 77, 1545–1554. doi:10.1246/bcsj.77.1545
Return to citation in text: [1] -
Suga, S.; Nishida, T.; Yamada, D.; Nagaki, A.; Yoshida, J. J. Am. Chem. Soc. 2004, 126, 14338–14339. doi:10.1021/ja0455704
Return to citation in text: [1] -
Maruyama, T.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 7324–7325. doi:10.1021/ja0511218
references cited therein.
Return to citation in text: [1] -
Okajima, M.; Suga, S.; Itami, K.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 6930–6931. doi:10.1021/ja050414y
Return to citation in text: [1] -
Lambert, J. B.; Kania, L.; Zhang, S. Chem. Rev. 1995, 95, 1191–1201. doi:10.1021/cr00037a003
Return to citation in text: [1] -
Olah, G. A.; Rasul, G.; Prakash, S. G. K. J. Am. Chem. Soc. 1999, 121, 9615–9617. doi:10.1021/ja9843167
Return to citation in text: [1] -
Lambert, J. B.; Yao, Z.; Zhang, M. J. Phys. Org. Chem. 2001, 14, 370–379. doi:10.1002/poc.377
Return to citation in text: [1] -
Kim, K. C.; Reed, C. A.; Elliott, D. W.; Mueller, L. J.; Rham, F.; Lin, L.; Lambert, J. B. Science 2002, 297, 825–827. doi:10.1126/science.1073540
Return to citation in text: [1] -
Okazaki, T.; Laali, K. K. J. Org. Chem. 2003, 68, 1827–1833. doi:10.1021/jo026312x
references therein.
Return to citation in text: [1] -
Bockholt, A.; Braun, T.; Jutzi, P.; Neumann, B.; Stammler, A.; Stammler, H. G. In Organosilicon Chemistry V; Auner, N.; Weis, J., Eds.; Wiley-VCH: Weinheim, 2003; pp 50–54.
Return to citation in text: [1] -
Kost, D.; Kingston, V.; Kalikhman, I. In Organosilicon Chemistry V; Auner, N.; Weis, J., Eds.; Wiley-VCH: Weinheim, 2003; pp 55–60.
Return to citation in text: [1] -
Kost, D.; Gostevskii, B.; Kocher, N.; Stalke, D.; Kalikhman, I. Angew. Chem., Int. Ed. 2003, 42, 1023–1026. doi:10.1002/anie.200390262
Return to citation in text: [1] -
Kalikhman, I.; Gostevskii, B.; Girshberg, O.; Sivaramakrishna, A.; Kocher, N.; Stalke, D.; Kost, D. J. Organomet. Chem. 2003, 686, 202–214. doi:10.1016/S0022-328X(03)00550-3
Return to citation in text: [1] -
Gostevskii, B.; Pestunovich, V.; Kalikhman, I.; Sivaramakrishna, A.; Kocher, N.; Deuerlein, S.; Leusser, D.; Stalke, D.; Kost, D. Organometallics 2004, 23, 4346–4348. doi:10.1021/om049575l
Return to citation in text: [1] -
Gostevskii, B.; Silbert, G.; Adear, K.; Sivaramakrishna, A.; Stalke, D.; Deuerlein, S.; Kocher, N.; Voronkov, M. G.; Kalikhman, I.; Kost, D. Organometallics 2005, 24, 2913–2920. doi:10.1021/om0500568
Return to citation in text: [1] -
Hatanaka, Y.; Okada, S.; Minami, T.; Goto, M.; Shimada, K. Organometallics 2005, 24, 1053–1055. doi:10.1021/om040132r
Return to citation in text: [1] -
Zhuikov, V. V. Russ. J. Gen. Chem. 1997, 67, 975–976.
Return to citation in text: [1] -
Zhuikov, V. V. Russ. J. Gen. Chem. 1999, 69, 1906–1911.
Return to citation in text: [1] -
Zhuikov, V. V. Russ. J. Gen. Chem. 2000, 70, 943–948.
Return to citation in text: [1] -
Yoshida, J.; Izawa, M. J. Am. Chem. Soc. 1997, 119, 9361–9365. doi:10.1021/ja970899t
Return to citation in text: [1] -
Yoshida, J.; Suga, S.; Fuke, K.; Watanabe, M. Chem. Lett. 1999, 251–252. doi:10.1246/cl.1999.251
Return to citation in text: [1] -
Mochida, K.; Itani, A.; Yokoyama, M.; Tuchiya, T.; Worley, S. D.; Kochi, J. K. Bull. Chem. Soc. Jpn. 1985, 58, 2149–2150. doi:10.1246/bcsj.58.2149
Return to citation in text: [1] -
Fukuzumi, S.; Kitano, T.; Mochida, K. Chem. Lett. 1990, 1741–1744. doi:10.1246/cl.1990.1741
Return to citation in text: [1] -
Lambert, J. B.; Zhao, Y. Angew. Chem., Int. Ed. Engl. 1997, 36, 400–401. doi:10.1002/anie.199704001
Return to citation in text: [1] -
Kraka, E.; Sopsa, C. P.; Grafenstein, J.; Cremer, D. Chem. Phys. Lett. 1997, 279, 9–16. doi:10.1016/S0009-2614(97)00944-5
Return to citation in text: [1] -
Muller, T.; Zhao, Y.; Lambert, J. B. Organometallics 1998, 17, 278–280. doi:10.1021/om971003w
See also ref 3c.
Return to citation in text: [1] -
Yamaguchi, K. J. Mass Spectrom. 2003, 38, 473–490. doi:10.1002/jms.488
Return to citation in text: [1] -
90% yield means that 1.8 mole of 5d was obtained from 1 mole of 1d.
Return to citation in text: [1]
| 1. | Yoshida, J.; Suga, S. Chem.–Eur. J. 2002, 8, 2650–2658. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S |
| 2. | Suga, S.; Watanabe, M.; Yoshida, J. J. Am. Chem. Soc. 2002, 124, 14824–14825. doi:10.1021/ja028663z |
| 3. | Suga, S.; Suzuki, S.; Maruyama, T.; Yoshida, J. Bull. Chem. Soc. Jpn. 2004, 77, 1545–1554. doi:10.1246/bcsj.77.1545 |
| 4. | Suga, S.; Nishida, T.; Yamada, D.; Nagaki, A.; Yoshida, J. J. Am. Chem. Soc. 2004, 126, 14338–14339. doi:10.1021/ja0455704 |
| 5. |
Maruyama, T.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 7324–7325. doi:10.1021/ja0511218
references cited therein. |
| 19. | Zhuikov, V. V. Russ. J. Gen. Chem. 1997, 67, 975–976. |
| 20. | Zhuikov, V. V. Russ. J. Gen. Chem. 1999, 69, 1906–1911. |
| 21. | Zhuikov, V. V. Russ. J. Gen. Chem. 2000, 70, 943–948. |
| 12. | Bockholt, A.; Braun, T.; Jutzi, P.; Neumann, B.; Stammler, A.; Stammler, H. G. In Organosilicon Chemistry V; Auner, N.; Weis, J., Eds.; Wiley-VCH: Weinheim, 2003; pp 50–54. |
| 13. | Kost, D.; Kingston, V.; Kalikhman, I. In Organosilicon Chemistry V; Auner, N.; Weis, J., Eds.; Wiley-VCH: Weinheim, 2003; pp 55–60. |
| 14. | Kost, D.; Gostevskii, B.; Kocher, N.; Stalke, D.; Kalikhman, I. Angew. Chem., Int. Ed. 2003, 42, 1023–1026. doi:10.1002/anie.200390262 |
| 15. | Kalikhman, I.; Gostevskii, B.; Girshberg, O.; Sivaramakrishna, A.; Kocher, N.; Stalke, D.; Kost, D. J. Organomet. Chem. 2003, 686, 202–214. doi:10.1016/S0022-328X(03)00550-3 |
| 16. | Gostevskii, B.; Pestunovich, V.; Kalikhman, I.; Sivaramakrishna, A.; Kocher, N.; Deuerlein, S.; Leusser, D.; Stalke, D.; Kost, D. Organometallics 2004, 23, 4346–4348. doi:10.1021/om049575l |
| 17. | Gostevskii, B.; Silbert, G.; Adear, K.; Sivaramakrishna, A.; Stalke, D.; Deuerlein, S.; Kocher, N.; Voronkov, M. G.; Kalikhman, I.; Kost, D. Organometallics 2005, 24, 2913–2920. doi:10.1021/om0500568 |
| 18. | Hatanaka, Y.; Okada, S.; Minami, T.; Goto, M.; Shimada, K. Organometallics 2005, 24, 1053–1055. doi:10.1021/om040132r |
| 7. | Lambert, J. B.; Kania, L.; Zhang, S. Chem. Rev. 1995, 95, 1191–1201. doi:10.1021/cr00037a003 |
| 8. | Olah, G. A.; Rasul, G.; Prakash, S. G. K. J. Am. Chem. Soc. 1999, 121, 9615–9617. doi:10.1021/ja9843167 |
| 9. | Lambert, J. B.; Yao, Z.; Zhang, M. J. Phys. Org. Chem. 2001, 14, 370–379. doi:10.1002/poc.377 |
| 10. | Kim, K. C.; Reed, C. A.; Elliott, D. W.; Mueller, L. J.; Rham, F.; Lin, L.; Lambert, J. B. Science 2002, 297, 825–827. doi:10.1126/science.1073540 |
| 11. |
Okazaki, T.; Laali, K. K. J. Org. Chem. 2003, 68, 1827–1833. doi:10.1021/jo026312x
references therein. |
| 6. | Okajima, M.; Suga, S.; Itami, K.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 6930–6931. doi:10.1021/ja050414y |
| 26. | Lambert, J. B.; Zhao, Y. Angew. Chem., Int. Ed. Engl. 1997, 36, 400–401. doi:10.1002/anie.199704001 |
| 27. | Kraka, E.; Sopsa, C. P.; Grafenstein, J.; Cremer, D. Chem. Phys. Lett. 1997, 279, 9–16. doi:10.1016/S0009-2614(97)00944-5 |
| 28. |
Muller, T.; Zhao, Y.; Lambert, J. B. Organometallics 1998, 17, 278–280. doi:10.1021/om971003w
See also ref 3c. |
| 24. | Mochida, K.; Itani, A.; Yokoyama, M.; Tuchiya, T.; Worley, S. D.; Kochi, J. K. Bull. Chem. Soc. Jpn. 1985, 58, 2149–2150. doi:10.1246/bcsj.58.2149 |
| 25. | Fukuzumi, S.; Kitano, T.; Mochida, K. Chem. Lett. 1990, 1741–1744. doi:10.1246/cl.1990.1741 |
| 23. | Yoshida, J.; Suga, S.; Fuke, K.; Watanabe, M. Chem. Lett. 1999, 251–252. doi:10.1246/cl.1999.251 |
| 22. | Yoshida, J.; Izawa, M. J. Am. Chem. Soc. 1997, 119, 9361–9365. doi:10.1021/ja970899t |
© 2007 Nokami et al; licensee Beilstein-Institut
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)