Abstract
In recent years, cyclic silicon-stereogenic silanes were successfully employed as stereoinducers in transition metal-catalyzed asymmetric transformations as exemplified by (1) the hydrosilylation of alkenes constituting a chirality transfer from silicon to carbon and (2) the kinetic resolution of racemic mixtures of alcohols by dehydrogenative silicon-oxygen coupling. In this investigation, a cyclic and a structurally related acyclic silane with silicon-centered chirality were compared using the above-mentioned model reactions. The stereochemical outcome of these pairs of reactions was correlated with and rationalized by the current mechanistic pictures. An acyclic silicon-stereogenic silane is also capable of inducing excellent chirality transfer (ct) in a palladium-catalyzed intermolecular carbon-silicon bond formation yet silicon incorporated into a cyclic framework is required in the copper-catalyzed silicon-oxygen bond forming reaction.

Graphical Abstract
Findings
Within the last decade, several asymmetric transformations based on silicon-stereogenic reagents or substrates were revisited or invented. [1-4] Aside from the use of silicon-stereogenic chiral auxiliaries in substrate-controlled reactions, [5] a still limited number of remarkable stereoselective processes with a stereogenic silicon as the reactive site were reported, [6] namely the inter- [7] as well as intramolecular [8] chirality transfers from silicon to carbon. Moreover, we had demonstrated that chiral silanes resolve racemic mixtures of alcohols in a non-enzymatic, transition metal-catalyzed kinetic resolution. [9]
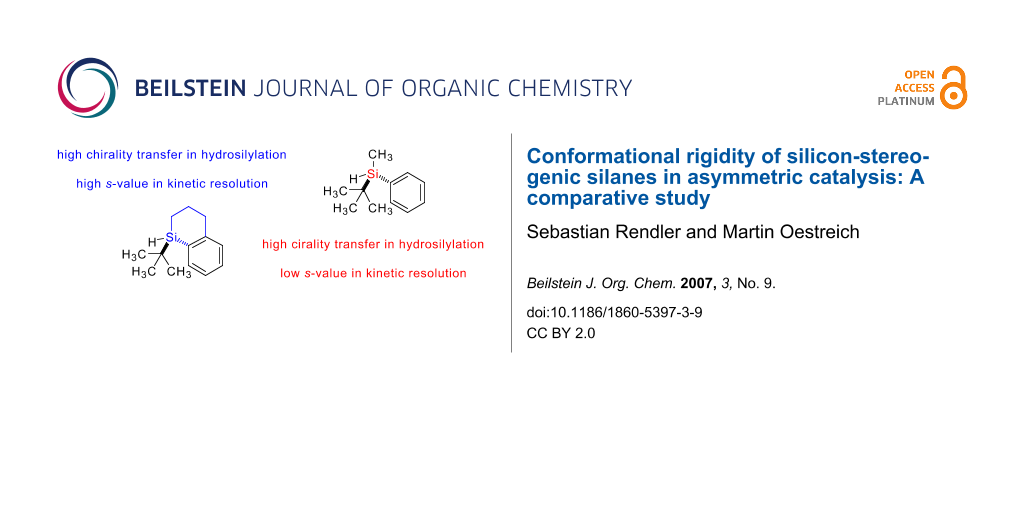
During our ongoing investigations directed towards the mechanistic elucidation of the origin of the chirality transfer in a palladium-catalyzed hydrosilylation, [10] we had to perform an extensive screening of silicon-stereogenic tertiary silanes. On that occasion, we became aware that a similar level of stereoselection was obtained when priveleged cyclic system 1a [11] was exchanged for the important acyclic congener 1b [12-15] (Figure 1). We had erroneously missed this known tertiary silane. This was particularly unfortunate in the light of the fact that these silanes are both decorated with three substituents of different steric demand and, therefore, display marked stereochemical differentiation around silicon.
![[1860-5397-3-9-1]](/bjoc/content/figures/1860-5397-3-9-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cyclic and acyclic sterically encumbered silanes.
Figure 1: Cyclic and acyclic sterically encumbered silanes.
In this preliminary communication, we wish to report a comparison of cyclic 1a and acyclic 1b as stereoinducers in the palladium-catalyzed chirality transfer from silicon to carbon and in the copper-catalyzed kinetic resolution of donor-functionalized alcohols capable of two-point binding.
The reagent-controlled hydrosilylation of norbornene derivative 2 with silane 1a proceeds with a perfect chirality transfer (rac-1a → rac-3a, Scheme 1). [8] Mechanistic investigation of the nature of the stereochemistry-determining step in this catalysis required a silane, which would produce slightly diminished diastereoselectivity and, hence, attenuated chirality transfer from silicon to carbon. [10] It was that situation that prompted us to investigate a considerable range of silicon-stereogenic silanes initially varied in ring size and exocyclic substituent; this was not met with satisfactory success. Based on the assumption that less rigid acyclic silanes would induce lower levels of diastereoselection, previously reported silane rac-1b – readily prepared in its racemic form [13] – was then supposed to serve such purpose. To our surprise, the palladium-catalyzed hydrosilylation of 2 with rac-1b gave almost perfect diastereoselectivity and good yield (rac-1b → rac-3b, Scheme 1).
![[1860-5397-3-9-i1]](/bjoc/content/inline/1860-5397-3-9-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cyclic and acyclic chiral silanes as potent reagents for the silicon-to-carbon chirality transfer.
Scheme 1: Cyclic and acyclic chiral silanes as potent reagents for the silicon-to-carbon chirality transfer.
This unexpected result inevitably introduced the pivotal question whether conformational rigidity of chiral silanes is a dispensible characteristic for asymmetric transformations. Thus, we subsequently tested rac-1b as resolving reagent in the kinetic resolution of an alcohol with a pending nitrogen donor (Scheme 2). In an earlier report, enantiomerically enriched silane 1a (96% ee) was applied in this diastereoselective copper-catalyzed dehydrogenative silicon-oxygen coupling affording promising optical purities for the unreacted alcohol ent-4 (84% ee) along with 5 (d.r. = 84:16) at 56% conversion. [9] For the present study, the diastereoselectivity of the formed ethers 5 is conclusive, which, in turn, allows for working with racemic silanes rac-1 (rac-1a → rac-5a versus rac-1b → rac-5b, Scheme 2). This is sufficient since the d.r. of 5 will be identical to the e.r. of the remaining alcohol 4 at exactly 50% conversion when using enantiopure silane 1. It must be noted that that diastereoselectivity is not dependent on conversion when using racemic silanes rac-1; conversely, using enantioenriched 1 it is.
![[1860-5397-3-9-i2]](/bjoc/content/inline/1860-5397-3-9-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Kinetic resolution of secondary alcohols using a dehydrogenative coupling reaction.
Scheme 2: Kinetic resolution of secondary alcohols using a dehydrogenative coupling reaction.
Whereas rac-5a was formed highly diastereoselectively (d.r. = 92:8) at 50% conversion, [9] the analogous reaction of rac-1b yielded rac-5b in a poor diastereomeric ratio (d.r. = 59:41) at comparable conversion. In sharp contrast to the results obtained in the hydrosilylation, embedding the asymmetrically substituted silicon into a cyclic framework appears to be an essential feature.
A comparison of the mechanisms of each reaction might serve as an explanation for this unexpected divergence. As outlined in Scheme 3, the hydrosilylation proceeds via a three-step catalytic cycle: (i) Reversible coordination of cationic silyl palladium species 6 by the alkene 2 (6 → 7), followed by (ii) fast and reversible migratory insertion forming β-silyl alkyl palladium intermediate 8 (7 → 8), and (iii) the involvement of a second silane moiety in the irreversible σ-bond metathesis. [10,16] Recent results clearly indicate step (ii) as diastereoselectivity-determining, revealing a thermodynamically controlled, reversible but highly diastereoselective migratory insertion step. [10]
![[1860-5397-3-9-i3]](/bjoc/content/inline/1860-5397-3-9-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Catalytic cycle for hydrosilylation.
Scheme 3: Catalytic cycle for hydrosilylation.
A different scenario might apply to the copper-catalyzed kinetic resolution of alcohols (Scheme 4). The phosphine-stabilized copper hydride 12 [17] is likely to be the catalytically active species, which is generated by alkoxide exchange (9 → 10) followed by a single catalytic turnover. The actual catalytic cycle then proceeds in a four-step propagation: (i) Coordination of pyridyl alcohol rac-4 accompanied by liberation of dihydrogen (12 → 10), (ii) rate-limiting dissociation of one phosphine ligand to generate a free coordination site, [18] (iii) coordination of the weakly donating chiral silane (10 → 11), followed by (iv) an exothermic and irreversible σ-bond metathesis [19] establishing the silicon-oxygen linkage in 5 and regenerating copper hydride 12 after coordination of another phosphine ligand (11 → 12). With steps (ii) and (iii) being reversible and chelate 10 being capable of alkoxide exchange, that is exchange of the optical antipodes of 4, one enantiomer of 4 is preferentially funnelled out via diastereomeric transition states (11 → 12).
![[1860-5397-3-9-i4]](/bjoc/content/inline/1860-5397-3-9-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Postulated catalytic cycle for dehydrogenative coupling.
Scheme 4: Postulated catalytic cycle for dehydrogenative coupling.
There is one major difference between the diastereoselectivity-determining steps in these catalytic cycles: (ii) in Scheme 3 and (iv) in Scheme 4. In the migratory insertion (ii, 7 → 8), carbon-silicon bond formation occurs between the stereogenic silicon and the prochiral carbon therefore entailing their close proximity. The newly formed stereogenic carbon is directly connected to the former source of chiral information. In contrast, the decisive asymmetrically substituted carbon atom in the alcohol substrate is more remote from the stereoselectivity-controlling silicon moiety in the silicon-oxygen bond formation (iv, 11 → 5). The stereogenic carbon in the alcohol is not directly involved in the actual bond formation. This mechanistic picture might account for the more demanding requirements to chiral silane 1: A cyclic framework leading to a locked conformation [11] improving the degree of organization in the stereochemistry-determining transition state 11.
In summary, we have shown for the first time that an excellent chirality transfer from silicon to carbon is also realized with suitably substituted acyclic silanes such as 1b. Our survey, however, underscores once more that cyclic silane 1a is a priveleged structure and certainly generally more applicable to catalytic asymmetric processes than 1b. The current mechanistic pictures provide a rather simple explanation for the observed stereochemical outcome of both diastereoselective carbon-silicon and silicon-oxygen bond formation. Based on this insight, further research will be devoted to the extension chiral silicon-based asymmetric catalysis.
Supporting Information
| Supporting Information File 1: Supporting Information. Experimental procedures and characterization data for all new compounds described in this manuscript. | ||
| Format: DOC | Size: 51.5 KB | Download |
Acknowledgements
The research was supported by the Deutsche Forschungsgemeinschaft (Emmy Noether program, Oe 249/2-3 and Oe 249/2-4), the Fonds der Chemischen Industrie (pre-doctoral fellowship to S. R.), and the Aventis Foundation (Karl Winnacker fellowship to M. O.). The authors thank Oliver Plefka for an orientating experiment. Generous donations of chemicals from Wacker AG (Burghausen/Germany) and Lanxess AG (Leverkusen/Germany) are gratefully acknowledged.
References
-
Maryanoff, C. A.; Maryanoff, B. E. In Synthesis and Utilization of Compounds with Chiral Silicon Centers. Asymmetric Synthesis; Morrison, J. D.; Scott, J. W., Eds.; Academic Press: Orlando, 1984; Vol. 4, pp 355–374.
Return to citation in text: [1] -
Sommer, L. H. Stereochemistry, Mechanism and Silicon; McGraw-Hill: New York, 1965.
Return to citation in text: [1] -
Sommer, L. H. Intra-Sci. Chem. Rep. 1973, 7, 1–44.
Return to citation in text: [1] -
Corriu, R. J. P.; Guérin, C.; Moreau, J. J. E. In Stereochemistry at Silicon. Topics in Stereochemistry; Eliel, E. L., Ed.; Wiley: New York, 1984; Vol. 15, pp 43–198.
Return to citation in text: [1] -
Bienz, S. Chimia 1997, 51, 133–139.
Return to citation in text: [1] -
Oestreich, M. Chem.–Eur. J. 2006, 12, 30–36. doi:10.1002/chem.200500782
Return to citation in text: [1] -
Schmidt, D. R.; O'Malley, S. J.; Leighton, J. L. J. Am. Chem. Soc. 2003, 12, 1190–1191. doi:10.1021/ja0283201
Return to citation in text: [1] -
Oestreich, M.; Rendler, S. Angew. Chem., Int. Ed. 2005, 44, 1661–1664. doi:10.1002/anie.200462355
Return to citation in text: [1] [2] -
Rendler, S.; Auer, G.; Oestreich, M. Angew. Chem., Int. Ed. 2005, 44, 7620–7624. doi:10.1002/anie.200502631
Return to citation in text: [1] [2] [3] -
Rendler, S.; Oestreich, M.; Butts, C. P.; Lloyd-Jones, G. C. J. Am. Chem. Soc. 2007, 129, 502–503. doi:10.1021/ja067780h
Return to citation in text: [1] [2] [3] [4] -
Rendler, S.; Auer, G.; Keller, M.; Oestreich, M. Adv. Synth. Catal. 2006, 348, 1171–1182. doi:10.1002/adsc.200606071
Return to citation in text: [1] [2] -
Bertrand, G.; Dubac, J.; Mazerolles, P.; Ancelle, J. Nouv. J. Chim. 1982, 6, 381–386.
Return to citation in text: [1] -
Larson, G. L.; Torres, E. J. Organomet. Chem. 1985, 293, 19–27. doi:10.1016/0022-328X(85)80241-2
Return to citation in text: [1] [2] -
Jankowski, P.; Schaumann, E.; Wicha, J.; Zarecki, A.; Adiwidjaja, G. Tetrahedron: Asymmetry 1999, 10, 519–526. doi:10.1016/S0957-4166(99)00022-1
Return to citation in text: [1] -
Jankowski, P.; Schaumann, E.; Wicha, J.; Zarecki, A.; Adiwidjaja, G.; Asztemborska, M. Chem. Commun. 2000, 1029–1030. doi:10.1039/b001440k
Return to citation in text: [1] -
LaPointe, A. M.; Rix, F. C.; Brookhart, M. J. Am. Chem. Soc. 1997, 119, 906–917. doi:10.1021/ja962979n
Return to citation in text: [1] -
Rendler, S.; Oestreich, M. Angew. Chem., Int. Ed. 2007, 46, 498–504. doi:10.1002/anie.200602668
Return to citation in text: [1] -
Rendler, S.; Oestreich, M. 2006. Unpublished results.
Return to citation in text: [1] -
Grimme, S.; Oestreich, M. 2006. Unpublished results.
Return to citation in text: [1]
| 17. | Rendler, S.; Oestreich, M. Angew. Chem., Int. Ed. 2007, 46, 498–504. doi:10.1002/anie.200602668 |
| 1. | Maryanoff, C. A.; Maryanoff, B. E. In Synthesis and Utilization of Compounds with Chiral Silicon Centers. Asymmetric Synthesis; Morrison, J. D.; Scott, J. W., Eds.; Academic Press: Orlando, 1984; Vol. 4, pp 355–374. |
| 2. | Sommer, L. H. Stereochemistry, Mechanism and Silicon; McGraw-Hill: New York, 1965. |
| 3. | Sommer, L. H. Intra-Sci. Chem. Rep. 1973, 7, 1–44. |
| 4. | Corriu, R. J. P.; Guérin, C.; Moreau, J. J. E. In Stereochemistry at Silicon. Topics in Stereochemistry; Eliel, E. L., Ed.; Wiley: New York, 1984; Vol. 15, pp 43–198. |
| 8. | Oestreich, M.; Rendler, S. Angew. Chem., Int. Ed. 2005, 44, 1661–1664. doi:10.1002/anie.200462355 |
| 10. | Rendler, S.; Oestreich, M.; Butts, C. P.; Lloyd-Jones, G. C. J. Am. Chem. Soc. 2007, 129, 502–503. doi:10.1021/ja067780h |
| 16. | LaPointe, A. M.; Rix, F. C.; Brookhart, M. J. Am. Chem. Soc. 1997, 119, 906–917. doi:10.1021/ja962979n |
| 7. | Schmidt, D. R.; O'Malley, S. J.; Leighton, J. L. J. Am. Chem. Soc. 2003, 12, 1190–1191. doi:10.1021/ja0283201 |
| 10. | Rendler, S.; Oestreich, M.; Butts, C. P.; Lloyd-Jones, G. C. J. Am. Chem. Soc. 2007, 129, 502–503. doi:10.1021/ja067780h |
| 9. | Rendler, S.; Auer, G.; Oestreich, M. Angew. Chem., Int. Ed. 2005, 44, 7620–7624. doi:10.1002/anie.200502631 |
| 9. | Rendler, S.; Auer, G.; Oestreich, M. Angew. Chem., Int. Ed. 2005, 44, 7620–7624. doi:10.1002/anie.200502631 |
| 12. | Bertrand, G.; Dubac, J.; Mazerolles, P.; Ancelle, J. Nouv. J. Chim. 1982, 6, 381–386. |
| 13. | Larson, G. L.; Torres, E. J. Organomet. Chem. 1985, 293, 19–27. doi:10.1016/0022-328X(85)80241-2 |
| 14. | Jankowski, P.; Schaumann, E.; Wicha, J.; Zarecki, A.; Adiwidjaja, G. Tetrahedron: Asymmetry 1999, 10, 519–526. doi:10.1016/S0957-4166(99)00022-1 |
| 15. | Jankowski, P.; Schaumann, E.; Wicha, J.; Zarecki, A.; Adiwidjaja, G.; Asztemborska, M. Chem. Commun. 2000, 1029–1030. doi:10.1039/b001440k |
| 10. | Rendler, S.; Oestreich, M.; Butts, C. P.; Lloyd-Jones, G. C. J. Am. Chem. Soc. 2007, 129, 502–503. doi:10.1021/ja067780h |
| 11. | Rendler, S.; Auer, G.; Keller, M.; Oestreich, M. Adv. Synth. Catal. 2006, 348, 1171–1182. doi:10.1002/adsc.200606071 |
| 13. | Larson, G. L.; Torres, E. J. Organomet. Chem. 1985, 293, 19–27. doi:10.1016/0022-328X(85)80241-2 |
| 10. | Rendler, S.; Oestreich, M.; Butts, C. P.; Lloyd-Jones, G. C. J. Am. Chem. Soc. 2007, 129, 502–503. doi:10.1021/ja067780h |
| 11. | Rendler, S.; Auer, G.; Keller, M.; Oestreich, M. Adv. Synth. Catal. 2006, 348, 1171–1182. doi:10.1002/adsc.200606071 |
| 9. | Rendler, S.; Auer, G.; Oestreich, M. Angew. Chem., Int. Ed. 2005, 44, 7620–7624. doi:10.1002/anie.200502631 |
| 8. | Oestreich, M.; Rendler, S. Angew. Chem., Int. Ed. 2005, 44, 1661–1664. doi:10.1002/anie.200462355 |
© 2007 Rendler and Oestreich; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)