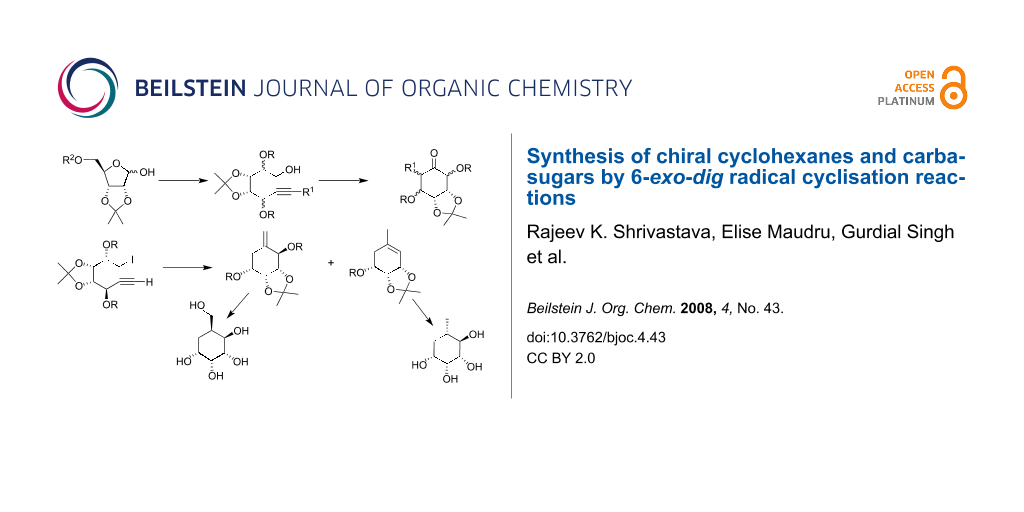
Abstract
Treatment of 5-(tert-butyldimethylsilyl)-2,3-O-isopropylidene-D-ribose with lithium acetylides gave mixtures of syn- and anti-alkynols 2a–2c which were separated following protection as methoxymethyl ethers. These were converted to the corresponding iodides which underwent 6-exo-dig radical cyclisation to afford chiral cyclohexanes and carbasugars. Oxidation of the primary alcohols 6a–b gave the corresponding aldehydes which on treatment with Grignard reagents afforded a mixture of alcohols. The corresponding iodides underwent similar 6-exo-dig cyclisation to give fully functionalised cyclohexanes.

Graphical Abstract
Introduction
The use of carbohydrates as precursors for highly functionalised carbocyclic rings systems has found wide utility in organic synthesis [1-11]. In particular there is a large amount of literature devoted to the synthesis of cyclopentanes [12,13] that employs a 5-exo-dig ring closure of a radical onto an alkyne [14]. In contrast there are fewer reports of the corresponding 6-exo-dig cyclisation to prepare fully functionalised cyclohexanes and carbasugars. The reason for this may be inferred from the observation that the formation of six-membered rings by a radical ring closure is some 40 times slower than the counterpart 5-hexenyl cyclisation [15-19]. In the case of the 6-hexenyl radical this results in the attack of the acyclic radical on the tin reagent becoming a more effective process compared to the 5-hexenyl case. An additional problem is that a 1,5-hydrogen abstraction leading to a resonance stabilised allylic radical is favourable, a reaction which may in fact be synthetically useful. We have previously reported the synthesis of cyclohexanes and carbasugars that uses a 6-exo-dig cyclisation [20] and herein we report on our current endeavours in this area.
Results and Discussion
Acetonisation [21] of D-ribose with acetone and hydrochloric acid afforded the corresponding acetonide (72%) which was converted into silyl ether 1 (83%) by treatment with tert-butyldiphenylsilyl chloride (TBDPSCl) and triethylamine using DMAP as the catalyst. The silylated D-riboses 1 were treated with lithium phenylacetylide to give an inseparable mixture of diastereoisomeric diols 2a in 89% yield (Scheme 1). Analysis of the 1H NMR spectrum showed a 1:2 isomeric mixture of diastereoisomers, on the basis of integration of the tert-butyl group resonances, which were slightly separated (syn: 1.08 ppm and anti: 1.09 ppm).
![[1860-5397-4-43-i1]](/bjoc/content/inline/1860-5397-4-43-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of iodides for radical cyclisation. Reagents and conditions: (i) LiC≡CPh, THF, −78 °C to RT 12 h; (ii) LiC≡CTMS, THF, −78 °C; (iii) MOMCl, iPr2NEt, DCM, 0 °C to RT 48 h; (iv) TBAF, THF, RT, 12 h; (v) I2, PPh3, Im, PhMe, Δ.
Scheme 1: Synthesis of iodides for radical cyclisation. Reagents and conditions: (i) LiC≡CPh, THF, −78 °C to ...
The stereochemistry at the new chiral centre was assigned on the basis of our previous findings [22]. The mixtures of diols 2 were treated with chloromethyl methyl ether to afford the corresponding MOM ethers, in a combined yield of 66%. At this juncture we were able to separate the diastereoisomers, 3a (anti) and 3b (syn), by flash column chromatography on silica gel. Alternatively the syn isomer 3b could be prepared almost exclusively using D-ribonolactone as the starting material [23].
Desilylation of 3a was effective with TBAF in THF and afforded the primary alcohol 6a in 87% yield, which was converted to the corresponding iodide 7a with triphenylphosphine, imidazole and iodine [24]. Similar chemistry with the isomer 3b gave the alcohol 6b and subsequently the iodide 7b in comparable yields.
With the availability of the iodides 7a and 7b we investigated their radical cyclisation reactions. Treatment of 7a with tri-n-butyltin hydride and AIBN in refluxing benzene afforded (Scheme 2) the 6-exo-cyclisation [25-28] products 10 in 93% yield as an inseparable mixture of E and Z geometric isomers in a ratio of 1:1.6 as determined by 1H NMR analysis. The diastereomer 7b on similar treatment underwent a 6-exo-cyclisation and afforded the corresponding cyclohexanes 11 and 12 in 61% yield. In this case we were able to separate the geometric E and Z isomers by silica gel chromatography, in a ratio of 3:2, respectively.
![[1860-5397-4-43-i2]](/bjoc/content/inline/1860-5397-4-43-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Radical cyclisation of compounds 7a and 7b.
Scheme 2: Radical cyclisation of compounds 7a and 7b.
The major isomer 11 was assigned having the Z geometry about the double bond on the basis of NOE experiments. Irradiation of the vinylic resonance at δ 6.66 resulted in enhancement of the aromatic resonances of H-6α at δ 2.93, and H-6β at δ 2.39. Analysis of the 1H NMR data (Table 1) allowed assignment of the stereochemistry at C-2. A large coupling constant J6α,5 11.8 Hz at δ 2.93 was observed, which suggests an axial-axial relationship between H-6α and H-5, whilst the resonance at δ 2.39, H-6β, showed a coupling constant J6β,5 5.3 Hz indicative of an axial-equatorial relationship.
Table 1: 270 MHz 1H NMR of compound 11.
| Proton | Chemical shift (ppm) | Multiplicity | Coupling constants (Hz) |
|---|---|---|---|
| 6β-H | 2.39 | dd | Jgem 12.5 and J6β,5 5.3 |
| 6α-H | 2.93 | td | Jgem 12.5 and J6α,5 11.8 |
| 5-H | 3.88 | dt | J5,6β 5.3, J5,6α 11.9 and J5,3 4.6 |
| 4-H | 4.20 | t | J4,5 = J4,3 5.3 |
| 3-H | 4.54 | t | J3,4 = J3,2 5.3 and J3,5 4.6 |
| 2-H | 4.61 | d | J2,3 5.3 |
| 7-H | 6.66 | s | |
Having been successful in our initial goal we turned our attention to the preparation of a cyclohexane that had an unsubstituted exo-methylene group. Treatment of the silylated ribose derivative with lithium trimethylsilylacetylide in THF gave the diols 2b, in a combined yield of 45% along with the alkynes 2c in 28% yield. The diastereomeric mixture 2b was converted to the MOM ethers 4a and 4b that could be separated in a combined yield of 64% with an anti:syn ratio of 3.5:1. Both of these diastereoisomers were processed separately. Removal of the silyl protecting group of 4a and 4b gave the corresponding primary alcohols 8a and 8b in 96% and 91% yields, respectively. These compounds were identical to those obtained from 2c after the diastereoisomers had been subjected to protection as MOM ethers on treatment with MOM chloride to give 5a and 5b followed by desilylation, the anti:syn ratio being 2.3:1.
The syn-product was correlated with the syn isomer 2c [23]. Conversion of the alcohols 8a and 8b to the iodides 9a and 9b was accomplished by treatment with triphenylphosphine, imidazole and iodine in 73% and 78% yields, respectively. At this point we proceeded to investigate the 6-exo radical cyclisation of these iodides. The syn-isomer 9b was treated with tri-n-butyltin hydride and AIBN in refluxing benzene, (Scheme 3) and gave the expected exo-methylene cyclohexane 13 in 38% yield along with the cyclohexene 14 in 49% yield where the OMOM group had been lost. The structure of 14 was clearly evident from its 1H NMR spectrum which had a resonance only for one MOM group in addition to the observation of a resonance at δ 5.33 due to the vinylic proton and a resonance at δ 1.73 due the methyl group.
![[1860-5397-4-43-i3]](/bjoc/content/inline/1860-5397-4-43-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Radical cyclisation of compound 9b.
Scheme 3: Radical cyclisation of compound 9b.
Following these findings we subjected the isomer 9a to the same reaction conditions and we observed that the cyclisation was appreciably slower, taking 24 h to go to completion. However this reaction was much more selective in that we only isolated 14 in a yield of 99%. In this case the primary radical formed from the iodide 9a undergoes a 6-exo cyclisation onto the alkyne, which results in the formation of a vinyl radical which is capable of abstracting a hydrogen from the methylene carbon of the OMOM group subsequently followed by β-scission [29,30] resulting in the formation of an allylic radical which gives rise to the observed product. The formation of 14 reflects the geometry of the vinyl radical and that these are bent with a bond angle of ca. 135° whilst in the case of an α-phenyl radical the geometry is linear [31]. In order to gain additional insight as to the geometries of the transition state(s) for these processes we undertook Monte Carlo calculations using much simpler structures (Figure 1), in order to simplify the computation, (18 and 19).
![[1860-5397-4-43-1]](/bjoc/content/figures/1860-5397-4-43-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of compounds 18 and 19.
Figure 1: Structures of compounds 18 and 19.
At low energies of 34.539 and 34.909 kcal/mol (Figure 2), the “trans” isomer 18 favours a boat conformation, in which the carbon-carbon double bond is above the plane of the ring and the methoxy group is in axial position and as a consequence the vinyl radical cannot abstract a hydrogen radical from the methoxy group. However at higher energies 35.657 kcal/mol, the “trans” isomer adopts a boat conformation and the ring can flip to different conformations at 35.786 and 35.931 kcal/mol and allows the radical to abstract a hydrogen atom from the methoxy group (Figure 2). The “cis” isomer 19 favours the chair conformation at low energies (36.007 kcal/mol) but can flip to the boat conformation more easily (36.394 kcal/mol) where the vinyl radical (Figure 3) can abstract the hydrogen atom from the methoxy group.
![[1860-5397-4-43-2]](/bjoc/content/figures/1860-5397-4-43-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Monte Carlo search on the simplified “trans” structures (structures shown are within 2 kcal/mol).
Figure 2: Monte Carlo search on the simplified “trans” structures (structures shown are within 2 kcal/mol).
![[1860-5397-4-43-3]](/bjoc/content/figures/1860-5397-4-43-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Monte Carlo search on the simplified “cis” structures (structures shown are within 2 kcal/mol).
Figure 3: Monte Carlo search on the simplified “cis” structures (structures shown are within 2 kcal/mol).
Having established that the 6-exo cyclisation was of synthetic value we proceeded to investigate the possibility of carrying out such a process using a secondary radical and thereby obtaining access to a cyclohexane ring that had substituents on all six carbons. Thus the alcohols 6a and 6b were oxidised most efficiently with PCC [32-37] to afford the corresponding aldehydes 20 and 21 in 85% and 90% yield, respectively. Both these aldehydes were found to be somewhat unstable at room temperature, as evidenced by their polymerisation and hence were used in the subsequent reaction as soon as possible. The aldehyde 20 was treated with phenylmagnesium chloride in THF and gave a mixture of two-diastereoisomeric alcohols 22 in 72% yield, which were separated using flash chromatography on silica gel. The stereochemistry at the newly created chiral centre could not be defined at this juncture. However, this was deemed to be unnecessary, as both of these diastereoisomeric alcohols would give rise to the same secondary radical and as a result we employed the diastereomeric mixture 22 in the subsequent chemistry. This mixture of alcohols 22 was converted to the corresponding iodides [25-30] 26 on treatment with triphenylphosphine, imidazole and iodine in 78% yield. The iodides 26 were prone to rapid decomposition even when stored at 4 °C as evidenced by TLC and 1H, 13C NMR analysis. Similar chemistry was performed with aldehyde 21 which gave a mixture of secondary alcohols 23 in 71% yield and subsequently the mixture of alcohol gave the corresponding iodide 27 in 73% yield. Similar treatment of the aldehydes 20 and 21 with methylmagnesium bromide afforded the analogous diols 24 and 25 in 80% and 79% yields, respectively; these diols were converted to the iodides 28 and 29 employing similar chemistry (Scheme 4).
![[1860-5397-4-43-i4]](/bjoc/content/inline/1860-5397-4-43-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of iodides from compounds 6a and 6b. Reagents and conditions: (i) PCC, 4 Å molecular sieves, DCM, RT, 2 h; (ii) PhMgCl or MeMgBr, THF, 0 °C, 1 h; (iii) PPh3, imidazole, I2, toluene, 60 °C, 2 h.
Scheme 4: Synthesis of iodides from compounds 6a and 6b. Reagents and conditions: (i) PCC, 4 Å molecular siev...
Radical cyclisation of the iodides 27 was conducted via dropwise addition of tri-n-butyltin hydride in refluxing benzene, using AIBN as a radical initiator. This resulted in the formation of the two pairs of diastereoisomers at C-2 and C-6 along with two pairs of geometric isomers in a combined yield of 91%, (Scheme 5). We were able to separate these isomers successfully by flash chromatography, eluting with light petroleum-diethyl ether (3:1 to 1:1). A similar outcome was obtained for the isomers 26 (Scheme 6) and resulted in a total cyclisation yield of 83%. We also investigated the cyclisation reactions of the “methyl” analogues 28 and 29 and these gave a similar outcome, (Scheme 6 and Scheme 5). Ozonolysis of the pair of geometric isomers 30 and 31 afforded the protected ketone 38 in 77% yield.
![[1860-5397-4-43-i5]](/bjoc/content/inline/1860-5397-4-43-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Radical cyclisation of compounds 27 and 29. Reagents and conditions: (i) Bu3SnH, AIBN, benzene, 80 °C, 3 h; (ii) DCM, MeOH, O3, −78 °C, 40 min.
Scheme 5: Radical cyclisation of compounds 27 and 29. Reagents and conditions: (i) Bu3SnH, AIBN, benzene, 80 ...
![[1860-5397-4-43-i6]](/bjoc/content/inline/1860-5397-4-43-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Radical cyclisation of compounds 26 and 28. Reagents and conditions: (i) Bu3SnH, AIBN, benzene, 80 °C, 3 h; (ii) DCM, MeOH, O3, −78 °C, 40 min.
Scheme 6: Radical cyclisation of compounds 26 and 28. Reagents and conditions: (i) Bu3SnH, AIBN, benzene, 80 ...
The diastereomeric ketone 39 was obtained in 74% yield after similar treatment of 32 and 33. Removal of the MOM and isopropylidene with 6 M HCl resulted in the aromatisation of the ring system. A similar outcome was obtained if we conducted acetylation of the crude reaction mixture in the presence of acetic anhydride and pyridine. In contrast to these findings the protecting groups of the cyclohexane 54, (Scheme 7), could be successfully removed on treatment using these conditions. Hydroboration of cyclohexene 14 obtained from cyclisation of 9a, following oxidative workup gave the cyclohexanol 54 in 77% yield. Removal of the isopropylidene and MOM protecting groups afforded a fully deprotected carba-β-D-rhamnose 55 in 99% yield. The structural integrity of 55 was established by acetylation with acetic anhydride and pyridine which gave the tetraacetate 56 in 99% yield.
![[1860-5397-4-43-i7]](/bjoc/content/inline/1860-5397-4-43-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of compounds 55 and 56.
Scheme 7: Synthesis of compounds 55 and 56.
Conclusion
We have established that carbohydrate derived alkynes undergo 6-exo radical cyclisation and provide access to fully functionalised cyclohexanes and carbasugars which are not easily accessible via other routes.
Supporting Information
| Supporting Information File 1: Experimental and Data | ||
| Format: DOC | Size: 280.5 KB | Download |
References
-
Ferrier, R. J.; Middleton, S. Chem. Rev. 1993, 93, 2779–2831. doi:10.1021/cr00024a008
Return to citation in text: [1] -
Arjona, O.; Gómez, A. M.; López, J. C.; Plumet, J. Chem. Rev. 2007, 107, 1919–2036. doi:10.1021/cr0203701
Return to citation in text: [1] -
McCasland, G. E.; Furuta, S.; Durham, L. J. J. Org. Chem. 1966, 31, 1516–1521. doi:10.1021/jo01343a048
Return to citation in text: [1] -
McCasland, G. E.; Furuta, S.; Durham, L. J. J. Org. Chem. 1968, 33, 2835–2841. doi:10.1021/jo01271a049
Return to citation in text: [1] -
McCasland, G. E.; Furuta, S.; Durham, L. J. J. Org. Chem. 1968, 33, 2841–2844. doi:10.1021/jo01271a050
Return to citation in text: [1] -
Lopez, J. C.; Fraser-Reid, B. Chem. Commun. 1997, 2251–2257. doi:10.1039/a704040g
Return to citation in text: [1] -
Tatsuta, K. J. Synth. Org. Chem., Jpn. 1997, 55, 970–981.
Return to citation in text: [1] -
Martínez-Grau, A.; Marco-Contelles, J. Chem. Soc. Rev. 1998, 27, 155–162. doi:10.1039/a827155z
Return to citation in text: [1] -
Dalko, P. I.; Sinaÿ, P. Angew. Chem., Int. Ed. 1999, 38, 773–777. doi:10.1002/(SICI)1521-3773(19990315)38:6<773::AID-ANIE773>3.0.CO;2-N
Return to citation in text: [1] -
Berecibar, A.; Grandjean, C.; Siriwardena, A. Chem. Rev. 1999, 99, 779–844. doi:10.1021/cr980033l
Return to citation in text: [1] -
Giese, B. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds; Pergamon Press: Oxford, 1986.
Return to citation in text: [1] -
Motherwell, W. B.; Crich, D. Free Radical Chain Reactions in Organic Synthesis; Academic Press: London, 1992.
Return to citation in text: [1] -
Giese, B.; Kopping, B.; Göbel, T.; Dickhaut, J.; Thoma, G.; Kulicke, K. J.; Trach, F. Org. React. 1996, 48, 301–856.
Return to citation in text: [1] -
McDevitt, R. E.; Fraser-Reid, B. J. Org. Chem. 1994, 59, 3250–3252. doi:10.1021/jo00091a004
Return to citation in text: [1] -
Beckwith, A. L. J. Tetrahedron 1981, 37, 3073–3100. doi:10.1016/S0040-4020(01)98839-8
Return to citation in text: [1] -
Beckwith, A. L. J.; Easton, C. J. J. Am. Chem. Soc. 1981, 103, 615–619. doi:10.1021/ja00393a019
Return to citation in text: [1] -
Malatesta, V.; Ingold, K. U. J. Am. Chem. Soc. 1981, 103, 609–614. doi:10.1021/ja00393a018
Return to citation in text: [1] -
Beckwith, A. L. J.; Schiesser, C. H. Tetrahedron 1985, 41, 3925–3941. doi:10.1016/S0040-4020(01)97174-1
Return to citation in text: [1] -
Jenkins, P. R.; Symons, M. C. R.; Booth, S. E.; Swain, C. J. Tetrahedron Lett. 1992, 33, 3543–3546. doi:10.1016/S0040-4039(00)92685-6
Return to citation in text: [1] -
Maudru, E.; Singh, G.; Wightman, R. W. Chem. Commun. 1998, 1505–1506. doi:10.1039/a802228c
Return to citation in text: [1] -
Hughes, N. A.; Speakman, P. R. H. Carbohydr. Res. 1965, 1, 171–175. doi:10.1016/S0008-6215(00)80921-0
Return to citation in text: [1] -
Mekki, B.; Singh, G.; Wightman, R. H. Tetrahedron Lett. 1991, 32, 5143–5146. doi:10.1016/S0040-4039(00)93450-6
Return to citation in text: [1] -
Jiang, S.; Singh, G.; Wightman, R. H. Chem. Lett. 1996, 67–68. doi:10.1246/cl.1996.67
Return to citation in text: [1] [2] -
Garegg, P. J.; Samuelsson, B. J. Chem. Soc., Perkin Trans. 1 1980, 2866–2869. doi:10.1039/P19800002866
Return to citation in text: [1] -
Gómez, A. M.; Danelón, G. O.; Moreno, E.; Valverde, S.; López, J. C. Chem. Commun. 1999, 175–176. doi:10.1039/a808848i
For recent 6-exo-dig radical cyclisations leading to carbasugars, see.
Return to citation in text: [1] [2] -
Gómez, A. M.; Moreno, E.; Valverde, S.; López, J. C. Tetrahedron Lett. 2002, 43, 7863–7866. doi:10.1016/S0040-4039(02)01919-6
Return to citation in text: [1] [2] -
Gómez, A. M.; Moreno, E.; Danelón, G. O.; Valverde, S.; López, J. C. Tetrahedron: Asymmetry 2003, 14, 2961–2974. doi:10.1016/S0957-4166(03)00532-9
Return to citation in text: [1] [2] -
Gómez, A. M.; Moreno, E.; Valverde, S.; López, J. C. Eur. J. Org. Chem. 2004, 1830–1840. doi:10.1002/ejoc.200300339
Return to citation in text: [1] [2] -
Rochigneux, I.; Fontanel, M.-L.; Malanda, J.-C.; Doutheau, A. Tetrahedron Lett. 1991, 32, 2017–2020. doi:10.1016/S0040-4039(00)78896-4
Return to citation in text: [1] [2] -
Malanda, J.-C.; Doutheau, A. J. Carbohydr. Chem. 1993, 12, 999–1016. doi:10.1080/07328309308020114
Return to citation in text: [1] [2] -
Nicolaides, A.; Borden, W. T. J. Am. Chem. Soc. 1991, 113, 6750–6755. doi:10.1021/ja00018a005
Return to citation in text: [1] -
Corey, E. J.; Suggs, J. W. Tetrahedron Lett. 1975, 2647–2650. doi:10.1016/S0040-4039(00)75204-X
Return to citation in text: [1] -
Corey, E. J.; Schmidt, G. Tetrahedron Lett. 1979, 399–402. doi:10.1016/S0040-4039(01)93515-4
Return to citation in text: [1] -
Omura, K.; Swern, D. Tetrahedron 1978, 34, 1651–1660. doi:10.1016/0040-4020(78)80197-5
Return to citation in text: [1] -
Albright, J. D.; Goldman, L. J. Am. Chem. Soc. 1965, 87, 4214–4216. doi:10.1021/ja01096a055
Return to citation in text: [1] -
Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070
Return to citation in text: [1]
| 1. | Ferrier, R. J.; Middleton, S. Chem. Rev. 1993, 93, 2779–2831. doi:10.1021/cr00024a008 |
| 2. | Arjona, O.; Gómez, A. M.; López, J. C.; Plumet, J. Chem. Rev. 2007, 107, 1919–2036. doi:10.1021/cr0203701 |
| 3. | McCasland, G. E.; Furuta, S.; Durham, L. J. J. Org. Chem. 1966, 31, 1516–1521. doi:10.1021/jo01343a048 |
| 4. | McCasland, G. E.; Furuta, S.; Durham, L. J. J. Org. Chem. 1968, 33, 2835–2841. doi:10.1021/jo01271a049 |
| 5. | McCasland, G. E.; Furuta, S.; Durham, L. J. J. Org. Chem. 1968, 33, 2841–2844. doi:10.1021/jo01271a050 |
| 6. | Lopez, J. C.; Fraser-Reid, B. Chem. Commun. 1997, 2251–2257. doi:10.1039/a704040g |
| 7. | Tatsuta, K. J. Synth. Org. Chem., Jpn. 1997, 55, 970–981. |
| 8. | Martínez-Grau, A.; Marco-Contelles, J. Chem. Soc. Rev. 1998, 27, 155–162. doi:10.1039/a827155z |
| 9. | Dalko, P. I.; Sinaÿ, P. Angew. Chem., Int. Ed. 1999, 38, 773–777. doi:10.1002/(SICI)1521-3773(19990315)38:6<773::AID-ANIE773>3.0.CO;2-N |
| 10. | Berecibar, A.; Grandjean, C.; Siriwardena, A. Chem. Rev. 1999, 99, 779–844. doi:10.1021/cr980033l |
| 11. | Giese, B. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds; Pergamon Press: Oxford, 1986. |
| 20. | Maudru, E.; Singh, G.; Wightman, R. W. Chem. Commun. 1998, 1505–1506. doi:10.1039/a802228c |
| 25. |
Gómez, A. M.; Danelón, G. O.; Moreno, E.; Valverde, S.; López, J. C. Chem. Commun. 1999, 175–176. doi:10.1039/a808848i
For recent 6-exo-dig radical cyclisations leading to carbasugars, see. |
| 26. | Gómez, A. M.; Moreno, E.; Valverde, S.; López, J. C. Tetrahedron Lett. 2002, 43, 7863–7866. doi:10.1016/S0040-4039(02)01919-6 |
| 27. | Gómez, A. M.; Moreno, E.; Danelón, G. O.; Valverde, S.; López, J. C. Tetrahedron: Asymmetry 2003, 14, 2961–2974. doi:10.1016/S0957-4166(03)00532-9 |
| 28. | Gómez, A. M.; Moreno, E.; Valverde, S.; López, J. C. Eur. J. Org. Chem. 2004, 1830–1840. doi:10.1002/ejoc.200300339 |
| 29. | Rochigneux, I.; Fontanel, M.-L.; Malanda, J.-C.; Doutheau, A. Tetrahedron Lett. 1991, 32, 2017–2020. doi:10.1016/S0040-4039(00)78896-4 |
| 30. | Malanda, J.-C.; Doutheau, A. J. Carbohydr. Chem. 1993, 12, 999–1016. doi:10.1080/07328309308020114 |
| 15. | Beckwith, A. L. J. Tetrahedron 1981, 37, 3073–3100. doi:10.1016/S0040-4020(01)98839-8 |
| 16. | Beckwith, A. L. J.; Easton, C. J. J. Am. Chem. Soc. 1981, 103, 615–619. doi:10.1021/ja00393a019 |
| 17. | Malatesta, V.; Ingold, K. U. J. Am. Chem. Soc. 1981, 103, 609–614. doi:10.1021/ja00393a018 |
| 18. | Beckwith, A. L. J.; Schiesser, C. H. Tetrahedron 1985, 41, 3925–3941. doi:10.1016/S0040-4020(01)97174-1 |
| 19. | Jenkins, P. R.; Symons, M. C. R.; Booth, S. E.; Swain, C. J. Tetrahedron Lett. 1992, 33, 3543–3546. doi:10.1016/S0040-4039(00)92685-6 |
| 14. | McDevitt, R. E.; Fraser-Reid, B. J. Org. Chem. 1994, 59, 3250–3252. doi:10.1021/jo00091a004 |
| 31. | Nicolaides, A.; Borden, W. T. J. Am. Chem. Soc. 1991, 113, 6750–6755. doi:10.1021/ja00018a005 |
| 12. | Motherwell, W. B.; Crich, D. Free Radical Chain Reactions in Organic Synthesis; Academic Press: London, 1992. |
| 13. | Giese, B.; Kopping, B.; Göbel, T.; Dickhaut, J.; Thoma, G.; Kulicke, K. J.; Trach, F. Org. React. 1996, 48, 301–856. |
| 32. | Corey, E. J.; Suggs, J. W. Tetrahedron Lett. 1975, 2647–2650. doi:10.1016/S0040-4039(00)75204-X |
| 33. | Corey, E. J.; Schmidt, G. Tetrahedron Lett. 1979, 399–402. doi:10.1016/S0040-4039(01)93515-4 |
| 34. | Omura, K.; Swern, D. Tetrahedron 1978, 34, 1651–1660. doi:10.1016/0040-4020(78)80197-5 |
| 35. | Albright, J. D.; Goldman, L. J. Am. Chem. Soc. 1965, 87, 4214–4216. doi:10.1021/ja01096a055 |
| 36. | Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041 |
| 37. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070 |
| 24. | Garegg, P. J.; Samuelsson, B. J. Chem. Soc., Perkin Trans. 1 1980, 2866–2869. doi:10.1039/P19800002866 |
| 23. | Jiang, S.; Singh, G.; Wightman, R. H. Chem. Lett. 1996, 67–68. doi:10.1246/cl.1996.67 |
| 23. | Jiang, S.; Singh, G.; Wightman, R. H. Chem. Lett. 1996, 67–68. doi:10.1246/cl.1996.67 |
| 29. | Rochigneux, I.; Fontanel, M.-L.; Malanda, J.-C.; Doutheau, A. Tetrahedron Lett. 1991, 32, 2017–2020. doi:10.1016/S0040-4039(00)78896-4 |
| 30. | Malanda, J.-C.; Doutheau, A. J. Carbohydr. Chem. 1993, 12, 999–1016. doi:10.1080/07328309308020114 |
| 22. | Mekki, B.; Singh, G.; Wightman, R. H. Tetrahedron Lett. 1991, 32, 5143–5146. doi:10.1016/S0040-4039(00)93450-6 |
| 21. | Hughes, N. A.; Speakman, P. R. H. Carbohydr. Res. 1965, 1, 171–175. doi:10.1016/S0008-6215(00)80921-0 |
| 25. |
Gómez, A. M.; Danelón, G. O.; Moreno, E.; Valverde, S.; López, J. C. Chem. Commun. 1999, 175–176. doi:10.1039/a808848i
For recent 6-exo-dig radical cyclisations leading to carbasugars, see. |
| 26. | Gómez, A. M.; Moreno, E.; Valverde, S.; López, J. C. Tetrahedron Lett. 2002, 43, 7863–7866. doi:10.1016/S0040-4039(02)01919-6 |
| 27. | Gómez, A. M.; Moreno, E.; Danelón, G. O.; Valverde, S.; López, J. C. Tetrahedron: Asymmetry 2003, 14, 2961–2974. doi:10.1016/S0957-4166(03)00532-9 |
| 28. | Gómez, A. M.; Moreno, E.; Valverde, S.; López, J. C. Eur. J. Org. Chem. 2004, 1830–1840. doi:10.1002/ejoc.200300339 |
© 2008 Shrivastava et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)