Abstract
Synthetic investigations towards the structurally complex and highly decorated framework of B-seco limonoid natural products by means of a [3,3]-sigmatropic rearrangement are described. Detailed model studies reveal, that an Ireland–Claisen rearrangement can be employed to construct the central C9–C10 bond thereby giving access to the B-seco limonoid scaffold. However, application of the developed strategy ended up failing in more complex and sterically demanding systems.

Graphical Abstract
Introduction
B-seco limonoids are a family of more than 100 highly oxygenated plant tetranortriterpenoids derived from the 4,4,8-trimethyl-17-furanylsteroid core structure I (Figure 1) [1-3]. Members of this natural product class, like 21-hydroxytoonacilide (1) [4,5] and prieurianin (2) [6-16] display antifeedant [6,7,17-21], antimalaria and anticancer [10,11,22-25] as well as diverse further bioactivities. Recently it was discovered that prieurianin (2) impairs the actin cytoskeleton by a mechanism that does not involve direct interaction with actin suggesting that its mode of action differs from previously known modulators [26].
![[1860-5397-10-15-1]](/bjoc/content/figures/1860-5397-10-15-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of the 4,4,8-trimethyl-17-furanylsteroid core structure I and the representative B-seco limonoids 21-hydroxytoonacilide (1) and prieurianin (2).
Figure 1: Structures of the 4,4,8-trimethyl-17-furanylsteroid core structure I and the representative B-seco ...
B-seco limonoids constitute exceptionally challenging synthesis targets, as the characteristic structural features are a compact, highly oxygenated as well as richly decorated framework and stereochemically dense functionalization. In all B-seco limonoids an A ring is linked by a C–C bond to a trans-fused bicyclic C–D ring having an exo-methylene moiety. The crowded C9–C10 bond bridging the two domains is the main synthetic obstacle.
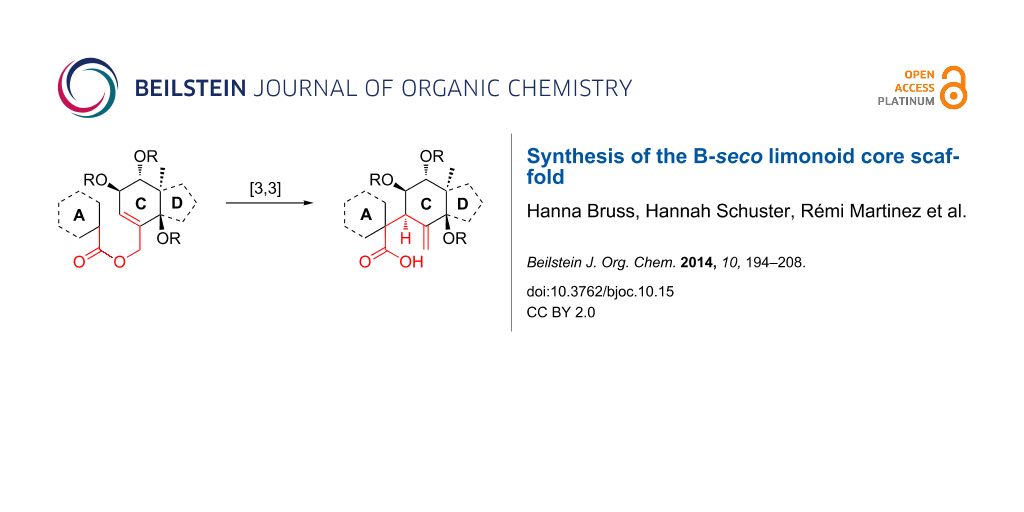
Taking into account the biology-oriented synthesis (BIOS) concept [27-34], which employs the scaffolds of biologically relevant natural product classes to inspire the synthesis of probes and reagents for chemical biology and medicinal chemistry research, we aimed at the development of a synthetic strategy to get access to the B-seco limonoid scaffold by means of a [3,3]-sigmatropic rearrangement as key step enabling the formation of the crucial C9–C10 bond (Scheme 1) [35]. In this paper we present a full report on this synthesis [36] as well as further synthetic studies towards the application of the developed strategy to the total synthesis of B-seco limonoid natural products.
![[1860-5397-10-15-i1]](/bjoc/content/inline/1860-5397-10-15-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of the B-seco limonoid framework employing a [3,3]-sigmatropic rearrangement for formation of the C9–C10 bond. R = Me or CO2H, LG = leaving group.
Scheme 1: Retrosynthetic analysis of the B-seco limonoid framework employing a [3,3]-sigmatropic rearrangemen...
Results and Discussion
Retrosynthetic analysis: Claisen rearrangement. In planning the synthesis we were inspired by Ley’s synthesis of azadirachtin in which a Claisen rearrangement has been successfully employed as key transformation [37,38]. Thus the allyl vinyl ether rearrangement precursor 11 was thought to be obtained from an O-alkylation between the thermodynamic enolate of 2-methylcyclohexanone (5) and the bicyclic electrophile 12 (Scheme 2). A challenging synthetic problem appears to be the construction of the stereochemically dense trans-fused C–D ring system 12, which possesses four stereogenic centers including two contiguous asymmetric quaternary centers at the ring junction. We decided to start the sequence with known enone 15 [39] and intended to construct the all-carbon quaternary center at C13 by substrate controlled α-functionalization. The second quaternary center at C14 might be established by 1,2-addition and finally, ring-closing metathesis would give rise to bicyclic system 12.
![[1860-5397-10-15-i2]](/bjoc/content/inline/1860-5397-10-15-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic analysis of the B-seco limonoid scaffold employing a Claisen rearrangement as key step for formation of the C9–C10 bond. PG = protecting group, LG = leaving group.
Scheme 2: Retrosynthetic analysis of the B-seco limonoid scaffold employing a Claisen rearrangement as key st...
Model studies towards the Claisen rearrangement. As the stereochemical substitution on the C ring system will have a major impact on the face selectivity of the planned Claisen rearrangement we defined precursors 19, 20 and 22 (Scheme 3) as suitable model systems, presenting appropriate stereogenic substitution at the C ring system. After merging these alcohols with an undecorated A ring, the resulting allyl vinyl ethers could serve as suitable rearrangement precursors.
![[1860-5397-10-15-i3]](/bjoc/content/inline/1860-5397-10-15-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of alcohols 19, 20 and 22. Reagents and conditions: a) CSA, 2,3-butanedione, trimethyl orthoformate, MeOH, reflux, 16 h, 93%; b) NaBH4, MeOH, rt, 0.5 h, quant.; c) silica-gel supported NaIO4, CH2Cl2/MeOH (20:1), rt, 3 h, quant.; d) MsCl, NEt3, CH2Cl2, 0 °C to rt, 3 h, quant.; e) paraformaldehyde, imidazole, THF/1 M NaHCO3 (1:1), rt, 2.5 h, 74%; f) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 15 min, quant.; g) LiHMDS, MeI, THF/DMPU (5:1), −78 °C to −10 °C, 91%, de = 100%; h) NaBH4, CeCl3·7H2O, MeOH, 0 °C, 15 min, 90%, de = 100%; i) PPh3, p-nitrobenzoic acid, DEAD, toluene, rt, 18 h; j) MeOH, Et2O, aqueous saturated K2CO3 solution, rt, 1 h, 64% (2 steps); for the synthesis of 19: k) DIPEA, MOMCl, CH2Cl2, reflux, 16 h, 99%; l) TBAF, THF, rt, 20 min, 94%; for the synthesis of 20: k) TIPSOTf, 2,6-lutidine, DMF, 0 °C to rt, 3 h, quant.; l) PTSA, MeOH, THF, rt, 30 h, 77%; m) PivCl, DMAP, pyridine, −15 °C to rt, 2 h, 77%; n) NaBH4, CeCl3·7H2O, MeOH, 0 °C to rt, 30 min, 96%, de = 100%; o) TFA, H2O, rt, 5 min, 72%; p) DIPEA, MOMCl, CH2Cl2, reflux, 15 h, 62%; q) DIBAL-H, CH2Cl2, −78 °C to rt, 2 h, quant.
Scheme 3: Synthesis of alcohols 19, 20 and 22. Reagents and conditions: a) CSA, 2,3-butanedione, trimethyl or...
The synthesis of 19 and 20 commenced with enone 15 [39], which was prepared from (−)-quinic acid (16) according to the route reported by Arthurs et al. [40] with minor modifications. After Baylis–Hillman reaction and subsequent silylation of the resulting free primary hydroxy group, substrate-controlled α-methylation of the lithium enolate proceeded with full stereocontrol [41,42], which can be explained by the strong conformational rigidity of the butane-2,3-diacetal (BDA) protected trans-diequatorial diols [43] and the stereoelectronic preference for axial attack on the electron-rich C13. Luche reduction with stereoelectronically preferred axial attack of the hydride gave alcohol 18 and Mitsunobu reaction installed the required stereochemistry at C14. The free C14 hydroxy group was masked with protecting groups (MOM and TIPS) of different size and chemical nature to examine the face-selectivity of the [3,3]-sigmatropic rearrangement. After selective desilylation, alcohols 19 and 20 were obtained.
In order to synthesize a model substrate without the rigid BDA-protecting group, compound 21 was treated with aqueous TFA to give a triol which was masked with three MOM-protecting groups (Scheme 3). Reductive cleavage of the pivaloyl group furnished alcohol 22.
Mesylation or tosylation of the primary alcohols in 19, 20 and 22 gave suitable electrophiles for the planned O-alkylation with the thermodynamic enolate of 2-methylcyclohexanone. However, under various conditions (NaH/15-crown-5/THF; t-BuOK/18-crown-6/THF/DMPU; KHMDS/THF) [44,45], the intended O-alkylation to yield the allyl vinyl ether failed. Equally, copper-catalyzed C–O couplings [46] of the alcohols 19, 20 and 22 with organotrifluoroborates failed or gave only low yields. Likewise Buchwald’s procedure for the copper-catalyzed coupling of primary alcohols with vinyl iodides to yield the allyl vinyl ether or, depending on the reaction conditions, directly the Claisen rearrangement products was not successful [47].
Alternative strategy: Ireland–Claisen rearrangement. As the Claisen rearrangement precursor, the allyl vinyl ether, could not be obtained under various conditions, we had to change the synthetic strategy and employed the Ireland variant of the Claisen rearrangement to construct the crucial C9–C10 bond (Scheme 4). In this strategy the A-ring 25 could be merged with the bicyclic C–D system 26 by esterification in order to obtain the allyl ester rearrangement precursor 24. Murai et al. [48] showed the utility of an Ireland–Claisen rearrangement in their model studies addressing the limonoid framework of azadirachtin.
![[1860-5397-10-15-i4]](/bjoc/content/inline/1860-5397-10-15-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Retrosynthetic analysis of the B-seco limonoid scaffold employing an Ireland–Claisen rearrangement as the key step for the formation of the C9–C10 bond. PG = protecting group.
Scheme 4: Retrosynthetic analysis of the B-seco limonoid scaffold employing an Ireland–Claisen rearrangement ...
Model studies towards the Ireland–Claisen rearrangement. To test the feasibility of this approach, model rearrangement precursor 27 was prepared from alcohol 19 (Scheme 5). Esterification with cyclohexanecarboxylic acid gave the desired allyl ester 27. Furthermore, to investigate the influence of the protecting groups at C11, C12 and C14 on the stereoselectivity and reaction rate, the allyl esters 28, 29 and 30 were prepared.
![[1860-5397-10-15-i5]](/bjoc/content/inline/1860-5397-10-15-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis and Ireland–Claisen rearrangement of the allyl esters 27, 28, 29 and 30. Reagents and conditions: a) DCC, DMAP, cyclohexanecarboxylic acid, CH2Cl2, rt, 4 h; b) TFA/H2O (3:1), rt, 30 min, 73% (2 steps); for the synthesis of 29: c) DIPEA, MOMCl, CH2Cl2, 40 °C, 15 h, 86%; for the synthesis of 30: c) imidazole, TBSCl, CH2Cl2, rt, 21 h, 45%; d) KHMDS, TMSCl, toluene, −78 °C to 85 °C, 18 h. Results: see Table 1.
Scheme 5: Synthesis and Ireland–Claisen rearrangement of the allyl esters 27, 28, 29 and 30. Reagents and con...
The four rearrangement precursors 27, 28, 29 and 30 were exposed to KHMDS and TMSCl in toluene, in order to induce the Ireland–Claisen rearrangement (Scheme 5, Table 1). The rigid BDA protecting group at C11 and C12 in 27 was not compatible with these conditions, resulting in only cleavage of the ester moiety (Table 1, entry 1). In case of the unprotected rearrangement precursor 28 an excess of base and TMSCl was used for in situ protection of the three free hydroxy groups as TMS ether, but the desired [3,3]-sigmatropic rearrangement could not be induced (Table 1, entry 2). However rearrangement of tris-MOM ether 29 as well as tris-TBS ether 30 gave the desired carboxylic acids 33 and 34 in high yield and excellent diastereoselectivity (Table 1, entries 3 and 4).
In view of the stereochemistry of the major diastereomers of the products, the rearrangement would occur from the re-face (transition state A, Figure 2). Thus, assuming the OTBS group in 30 at C14 is pseudo-axial to avoid allylic A1,2-strain, the sigmatropic rearrangement occurred via a pseudo-axial attack of the silyl ketene acetal on the double bond in the cyclohexene ring. These results are in accordance with the observations of Ireland et al. [49], who examined the propensity for axial versus equatorial attack in the rearrangement of cyclohexenyl acetates and observed a strong preference for the stereochemically controlled axial approach. The size of the protecting groups had a strong influence on the face-selectivity, furnishing in case of the TBS derivative only one diastereomer. By substitution of C14 with a bulky group (1,2-allylic strain) the ring inversion barrier of the cyclohexene conformations might be increased, resulting in higher de values.
![[1860-5397-10-15-2]](/bjoc/content/figures/1860-5397-10-15-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Conformation of rearrangement precursor 30 and possible transition state involved in the Ireland–Claisen rearrangement.
Figure 2: Conformation of rearrangement precursor 30 and possible transition state involved in the Ireland–Cl...
These model studies suggested that an Ireland–Claisen rearrangement is a feasible strategy to construct the C9–C10 bond in B-seco limonoids.
In order to perform further model studies, we established a straight forward synthetic access to model substrates without the BDA group (Scheme 6). With the TBDPS- instead of the TBS ether on the primary hydroxy group, the BDA group in alcohol 35 could be selectively cleaved by treatment with TFA in aqueous CH2Cl2 to release triol 36, which was masked with different protecting groups (MOM, TBS, Piv). After desilylation, the liberated alcohols 40, 41 and 42 could be esterified with various cyclic and acyclic model A rings to give the targeted rearrangement precursors A (Scheme 6, Table 2).
![[1860-5397-10-15-i6]](/bjoc/content/inline/1860-5397-10-15-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of model C rings 40, 41 and 42. Reagents and conditions: a) TBDPSCl, DMAP, NEt3, CH2Cl2, rt, 18 h, 93%; b) LiHMDS, MeI, THF/DMPU (10:1), −78 °C to 0 °C, 1.5 h, 98%; c) CeCl3·7H2O, NaBH4, MeOH, 0 °C, 15 min, 86%; d) Ph3P, p-nitrobenzoic acid, DEAD, toluene, 15 h; e) MeOH, Et2O, aqueous saturated K2CO3-solution, rt, 2 h, 85% (2 steps); f) CH2Cl2/TFA/H2O (2:1:0.1), rt, 10 min, 85%; for the synthesis of 37: g) DIPEA, MOMCl, NaI, THF, 65 °C, 4.5 h, 98%; for the synthesis of 38 g) TBSCl, imidazole, DMF, rt, 18 h, 92%; for the synthesis of 39: PivCl, DMAP, pyridine, rt, 4 d, 78%; for the synthesis of 40: h) 37, TBAF, THF, rt, 2 h, 95%; for the synthesis of 41: h) 38, 10% NaOH/MeOH, reflux, 6.5 h, 72%; for the synthesis of 42: h) 39, HF·pyridine, THF, rt, 24 h, 71%.
Scheme 6: Synthesis of model C rings 40, 41 and 42. Reagents and conditions: a) TBDPSCl, DMAP, NEt3, CH2Cl2, ...
Table 2: Esterification of alcohols 40, 41 and 42 and Ireland–Claisen rearrangement. Reagents and conditions: a) EDC·HCl, DMAP, carboxylic acid, CH2Cl2, rt, 15–22 h; for 52: silylation of the free hydroxy group at C14: imidazole, TBSCl, DMAP, DMF, rt, 40 h; b) KHMDS, TMSCl, toluene, −78 °C to 85 °C, ca. 18 h.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-15-i16.svg?max-width=637&scale=1.0)
|
|||
| entry | alcohol | allyl ester A | carboxylic acid B |
|---|---|---|---|
| 1 | 40 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-15-i17.svg?max-width=637&scale=1.0)
43 (87% yield) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-15-i18.svg?max-width=637&scale=1.0)
44 (75% yield, de = 80%) |
| 2 | 40 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-15-i19.svg?max-width=637&scale=1.0)
45 (81% yield) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-15-i20.svg?max-width=637&scale=1.0)
46 (93% yield, de = 78%) |
| 3 | 40 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-15-i21.svg?max-width=637&scale=1.0)
47 (93% yield) |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-15-i22.svg?max-width=637&scale=1.0)
48 (quant. yield, de = 72%) |
| 4 | 40 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-15-i23.svg?max-width=637&scale=1.0)
49 (quant. yield) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-15-i24.svg?max-width=637&scale=1.0)
50 (95% yield, de = 72%) |
| 5 | 41 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-15-i25.svg?max-width=637&scale=1.0)
51 (79% yield) |
– |
| 6 | 41 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-15-i26.svg?max-width=637&scale=1.0)
52 (78% yield, 2 steps) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-15-i27.svg?max-width=637&scale=1.0)
53 (quant. yield, de = 94%) |
| 7 | 42 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-15-i28.svg?max-width=637&scale=1.0)
54 (95% yield) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-15-i29.svg?max-width=637&scale=1.0)
55 (90% yield, de = 94%) |
The obtained rearrangement precursors were submitted to the Ireland–Claisen rearrangement conditions and gave the desired carboxylic acids B in excellent yield and diastereoselectivity (Scheme 6, Table 2). The presence of the keto functionality in 43 is compatible with the rearrangement conditions (Table 2, entry 1). Intermediary, the silyl enol ether and the silyl ketene acetal are formed. However, after the rearrangement, the keto-functionality can be set free again during an acidic work-up. In terms of yield and diastereoselectivity there was no difference observed between rearrangements with derivatives with a cyclic or acyclic model A ring. Rearrangement precursor 51 containing a free hydroxy group could not be converted into the desired carboxylic acid (Table 2, entry 5). Under the same conditions but by using an excess of base and TMSCl for the in situ protection of the free hydroxy group, only the starting material could be reisolated.
Encouraged by these findings we attempted to perform the [3,3]-sigmatropic rearrangement with C1-substituted A rings, as many B-seco limonoids are oxygenated at this position. For this purpose, both the anti- and syn-substituted β-alkoxy esters 56 and 57 were synthesized (Scheme 7; for experimental procedure see Supporting Information File 1). In the literature [50] not many examples are known in which β-alkoxy esters serve as rearrangement precursors as the β-elimination of the alkoxy group can easily occur under the rearrangement conditions. Indeed, under various conditions by using different bases (LDA, LiHMDS, KHMDS) and solvents (THF, toluene, THF/DMPU) and by carefully controlling the reaction conditions (deprotonation at low temperature (−100 °C), gradual warming of the reaction mixture), only the elimination product 58 could be isolated.
![[1860-5397-10-15-i7]](/bjoc/content/inline/1860-5397-10-15-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: β-Substituted allyl esters tested in the Ireland–Claisen and the Carroll rearrangement.
Scheme 7: β-Substituted allyl esters tested in the Ireland–Claisen and the Carroll rearrangement.
Furthermore, a dianionic Ireland–Claisen rearrangement employing β-hydroxy ester 59 was unsuccessful (Scheme 7). Under various conditions, we observed only decomposition of the starting material by cleavage of the ester moiety. Likewise, cleavage of the ester moiety occurred in an attempt to perform a Carroll rearrangement with β-keto ester 60.
These studies show that it might be necessary to oxygenate at C1 after the sigmatropic rearrangement because the C1 substitution seems to have major impact on the success of the rearrangement.
Synthesis of the B-seco limonoid scaffold. Encouraged by the results of the rearrangements of the model substrates with an undecorated A ring, we attempted to access the B-seco limonoid scaffold by an analogous rearrangement of bicyclic precursor 66 (Scheme 8).
![[1860-5397-10-15-i8]](/bjoc/content/inline/1860-5397-10-15-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis and Ireland–Claisen rearrangement of bicyclic allyl ester precursor 66. Reagents and conditions: a) LiHMDS, TMSCl, THF, −78 °C to 0 °C, 90 min; b) NBS, THF, 0 °C, 90 min, 68% (2 steps); c) allyltributyltin, AIBN, toluene, 80 °C, 18 h, 81%, de = 80%; d) KHMDS, THF, −78 °C, 1 h, then MeI, rt, 20 min, 74%, de = 100%; e) PdCl2(CH3CN)2, toluene, 65 °C, 2 days, 70% (81% brsm); f) paraformaldehyde, imidazole, THF/1 M NaHCO3 (2:1), rt, 19 h, 74%; g) TFA/H2O (3:1), rt, 30 min, 84%; h) TBSCl, imidazole, DMF, rt, 18 h, 94%; i) tetraallyltin, BuLi, THF, −78 °C to rt, 30 min, 72%, dr = ca. 2:1; j) Grubbs I, CH2Cl2, rt, 18 h, 80%; k) HF·pyridine, THF, 0 °C to rt, 20 h, 99%; l) EDC·HCl, 4-oxocyclohexanecarboxylic acid (25), DMAP, CH2Cl2, rt, 16 h, 80%; m) DIPEA, MOMCl, NaI, THF, 50 °C, 21 h, 62%; n) LiHMDS, supernatant of a centrifuged mixture of TMSCl/NEt3 (v/v = 1:1) and toluene, toluene, −78 °C to 65 °C in 6 h, 60 h at 65 °C, 88% yield, dr = 1:2 (67:68).
Scheme 8: Synthesis and Ireland–Claisen rearrangement of bicyclic allyl ester precursor 66. Reagents and cond...
The elaboration of the required bicyclic system commenced with enone 15 (Scheme 8). Initially, we envisaged an α-vinylation via Buchwald’s procedure for the catalytic asymmetric vinylation of enones [51]. However, the desired vinylated product could not be obtained under the described conditions. An alternative α-formylation/Wittig olefination sequence gave only low yields. O’Brien et al. [41] described the failure of a direct alkylation of the lithium enolate of 15 with alkyl halides under several conditions. They incorporated an α-allyl side chain via an α-bromo-enone, which can be obtained from an initially formed silyl enol ether, and subsequent reaction with NBS. Keck allylation of the α-bromo-enone using allyltributyltin and AIBN gave the desired α-allylated product 61. We used the high substrate control to construct the first quaternary center at C13 by trapping the potassium enolate of 61 with MeI, furnishing the desired product as a single diastereomer. Isomerization of the terminal double bond with cat. PdCl2(CH3CN)2 and Baylis–Hillman reaction proceeded uneventfully to afford compound 62. Deprotection of the butane-2,3-diacetal under acidic conditions followed by selective silylation of the primary and the allylic hydroxy groups gave alcohol 63.
The second quaternary center of the bicyclic C–D system 66 was envisaged to be constructed by 1,2-addition, using the free β-hydroxy functionality in 63 as directing group. Several conditions with allyl boronates, stannanes, silanes, indium, magnesium bromide, cerium, zinc bromide and other reagents have been screened. Finally, the best result was achieved with tetraallyltin and BuLi affording a 2:1 mixture of diastereomers, with the desired diastereomer being the minor product. The stereochemistry was unambiguously confirmed by crystal structure analysis of a derivative of the major diastereomer [36]. Thus, the preference for the axial attack predominates the aimed directing effect of the β-hydroxy group.
Bicyclic system 65 could be obtained by ring-closing metathesis using Grubbs 1st generation catalyst and subsequent selective deprotection of the primary silyl ether. After esterification with 4-oxocyclohexanecarboxylic acid (25) and protection of the remaining two free hydroxy groups as MOM ethers, which was accompanied by silyl migration, the synthesis of rearrangement precursor 66 was completed.
Application of the developed reaction conditions for the rearrangement of the model systems was fruitless, resulting in only cleavage of the ester moiety. Crucial for the success of the envisaged Ireland–Claisen rearrangement was a gradual warming of the reaction mixture from −78 °C to 65 °C over a period of 6 h and the addition of the supernatant of a centrifuged mixture of TMSCl, NEt3 and toluene instead of the addition of unactivated TMSCl. When 66 was exposed to TMSCl/NEt3 and LiHMDS in toluene, the intended rearrangement occurred smoothly giving rise to 67 and 68 (ca. 1:2 ratio, 88% combined yield) through the desired C9–C10 bond formation (Scheme 8). The configuration of the diastereomers was determined based on the analysis of the coupling constants of H9 and H11 and nOe signal enhancements.
In contrast to the high face-selectivity in the rearrangement of the model systems, the reaction seems to take place from both sides of the bicyclic C–D system. However, in this case the si-face approach appears to be favoured (transition state C, Figure 3a). This might be explained by the rigidity of the bicyclic system, such that transition states B and C can compete without preference for a conformation that clearly favours axial attack. Underscored by MM2 conformational calculations, it is furthermore plausible that the C11 oxygen can form an H-bond to the terminal methyl C–H of the C14 MOM group, thus blocking a pseudo-axial approach and leading to the observed reversal in selectivity.
![[1860-5397-10-15-3]](/bjoc/content/figures/1860-5397-10-15-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Conformations of rearrangement precursors 66 and 77 and possible transition states involved in the Ireland–Claisen rearrangements. R = MOM.
Figure 3: Conformations of rearrangement precursors 66 and 77 and possible transition states involved in the ...
We hypothesized that the diastereoselectivity could be improved by performing the rearrangement with the open-chain precursor 70 (Scheme 9), which should be less rigid than the bicyclic system 66 and therefore, in analogy to the results of the rearrangements with the model substrates, probably preferably rearrange via a pseudo-axial attack. The open-chain precursor 70 was obtained by MOM protection of diol 64, selective cleavage of the primary TBS group and subsequent esterification with 4-oxocyclohexanecarboxylic acid (25).
![[1860-5397-10-15-i9]](/bjoc/content/inline/1860-5397-10-15-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis and Ireland–Claisen rearrangement of allyl ester 70. Reagents and conditions: a) DIPEA, MOMCl, NaI, THF, 65 °C, 3 days, 35%; b) HF·pyridine, THF, 20 h, rt, 83%; c) EDC·HCl, DMAP, CH2Cl2, 4-oxocyclohexanecarboxylic acid (25), rt, 24 h, 83%; d) LiHMDS, TMSCl/NEt3, toluene, 1 h at −78 °C, then gradual warming to 65 °C within 6 h and 40 h at 65 °C, yield n.d. dr = 1:0; e) Grubbs I, CH2Cl2, rt, 20 h, yield n.d.
Scheme 9: Synthesis and Ireland–Claisen rearrangement of allyl ester 70. Reagents and conditions: a) DIPEA, M...
However, in this case the [3,3]-sigmatropic rearrangement proceeded exclusively via pseudo-equatorial attack, giving after RCM the C9-epi limonoid scaffold 68 as single diastereomer. The sterically demanding rearrangement precursor 70 seems to allow only the undesired pseudo-equatorial attack.
We envisaged that protecting the free secondary hydroxy group in 71 with a sterically demanding TIPS group might induce a conformational change of the rearrangement precursor and intended to investigate the influence of this conformational change on the diastereoselectivity of the Ireland–Claisen rearrangement (Scheme 10). However, the allyl ester 72 seems to be too sterically hindered to allow the [3,3]-sigmatropic rearrangement to proceed. After 2 days reaction time, only traces of rearrangement product 73 could be observed.
![[1860-5397-10-15-i10]](/bjoc/content/inline/1860-5397-10-15-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis and Ireland–Claisen rearrangement of allyl ester 72. Reagents and conditions: a) TIPSOTf, pyridine, DMAP, rt, 22 h, 60%; b) DIPEA, MOMCl, THF, 25 h, 50 °C, 29%; c) LiHMDS, TMSCl/NEt3, toluene, 1 h at −78 °C, then gradual warming to 65 °C within 6 h and stirred for 2 days at 65 °C.
Scheme 10: Synthesis and Ireland–Claisen rearrangement of allyl ester 72. Reagents and conditions: a) TIPSOTf,...
Moreover, in order to allow the synthetic access to further B-seco limonoid analogues, the C14-epi B-seco limonoid scaffold 78 and C14-epi/C9-epi scaffold 79 were accessed (Scheme 11). Starting from diol 74, the C14-epi rearrangement precursor 77 was synthesized employing a sequence of ring-closing metathesis, TBS deprotection, esterification and MOM protection. The Ireland–Claisen rearrangement proceeded smoothly and gave a ca. 1.3:1 (78:79) mixture of diastereomers with the product resulting from the pseudo-axial attack of the silyl ketene acetal being the major diastereomer.
![[1860-5397-10-15-i11]](/bjoc/content/inline/1860-5397-10-15-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of the C14-epi and C14/C9-epi B-seco limonoid scaffolds 78 and 79. Reagents and conditions: a) Grubbs I, CH2Cl2, rt, 18 h, 86%; b) DIPEA, MOMCl, NaI, THF, 20 h, 65 °C, 89%; c) HF·pyridine, THF, 0 °C to rt, 24 h, 76%; d) EDC·HCl, DMAP, CH2Cl2, 4-oxocyclohexanecarboxylic acid (25), rt, 19 h, 92%; e) LiHMDS, TMSCl/NEt3, toluene, 1 h at −78 °C, then gradual warming to 65 °C within 6 h and stirred for 43 h at 65 °C, 78% yield, dr = 1.3:1 (78:79).
Scheme 11: Synthesis of the C14-epi and C14/C9-epi B-seco limonoid scaffolds 78 and 79. Reagents and condition...
In analogy to the results above the transition states D and E seem to compete without any preference for a conformation that clearly favours axial attack (Figure 3b). Presumably because of the absence of electrostatic interactions in the pseudo-axial attack of the silyl ketene acetal (compare transition state B in Figure 3a: electrostatic interaction with -OMOM at C14), this approach is slightly favored.
Synthesis of fully functionalized A ring. Having these extensive studies completed we then focused on the synthesis of a suitable fully functionalized A ring 87 (Scheme 12) that after connection to the bicyclic C–D system and subsequent Ireland–Claisen rearrangement was supposed to give access to the entire framework of B-seco limonoids.
![[1860-5397-10-15-i12]](/bjoc/content/inline/1860-5397-10-15-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of fully functionalized A ring 87. Reagents and conditions: a) HO(CH2)2OH, THF, Pd/C, H2, pH 5, rt, overnight, 97%, de = 100%; b) LDA, TMSCl, THF, −78 °C to rt, 1.5 h, quant.; c) O3, CH2Cl2, −78 °C, then DMS, −78 °C to rt; d) TMSCHN2, CH2Cl2/MeOH (1:1), rt, 0.5 h, 59% (2 steps); e) NaBH4, MeOH, 0 °C, 0.5 h; f) TBDPSCl, imidazole, DMAP, CH2Cl2, 0.5 h, rt, 78% (2 steps); g) CH2Cl2/H2O/HClO4 (25:5:1), rt, 6 h; h) LiHMDS, TMSCl, THF, −78 °C to rt, 1.5 h; i) Pd(OAc)2, DMSO, O2, overnight, 72% (3 steps); j) LDA, 1H-benzotriazole-1-methanol, THF, −78 °C, 3 h, 67%, de = 100%; k) TBSCl, imidazole, DMF, rt, overnight, 90%; l) H2O2, NaOH, MeOH, 0 °C, 1 h, 80%, de = 100%; m) NaBH4, (PhSe)2, EtOH, 0 °C to rt, 10 min, 93%; n) TESCl, imidazole, DMF, 40 °C, 2 h, 99%; o) NaBH4, MeOH, 0 °C, 3 h, 64%; p) LiBH4, THF, 65 °C, 4 days, 67% (92% brsm); q) MOMCl, DIPEA, NaI, THF, 50 °C, 4 days, 96%, dr = 2:1; r) 5% TFA in CH2Cl2, rt, 0.5 h, 74%; s) DMP, NaHCO3, CH2Cl2, 0 °C, 3 h, 76%; t) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O (4:1), rt, 3.5 h, 91%.
Scheme 12: Synthesis of fully functionalized A ring 87. Reagents and conditions: a) HO(CH2)2OH, THF, Pd/C, H2,...
Explorations to access this fragment started from Hajos–Parrish ketone 80 [52] (Scheme 12), as we were inspired by a reaction sequence Arseniyadis et al. [53] used in their synthesis of a left-half taxoid building block. Regioselective protection of the less hindered ketone in 80 and diastereoselective hydrogenation of the double bond could be achieved in a known one-pot procedure [54,55] affording the cis-hydrindanone and providing the desired stereochemistry at C4 and C5. Ozonolytic cleavage of the corresponding silyl enol ether 81 followed by esterification with TMSCHN2 furnished the ester aldehyde that was reduced to the primary alcohol and protected to give TBDPS ether 82. After selective cleavage of the acetal group by treatment with perchloric acid, installation of the double bond via Saegusa oxidation [56,57] of the previously formed TMS ether furnished enone 83 as the major regioisomer. Minor amounts of the undesired regioisomer could be separated by column chromatography. Hydroxymethylation of 83 using 1H-benzotriazole-1-methanol proceeded diastereoselectively due to substrate control [58]. The configuration was determined by the high coupling constant (JH4/H5 = 12.6 Hz) indicating the trans-diaxial orientation of H4 and H5. Silylation of the primary hydroxy group afforded compound 84 that was converted into the epoxide with complete stereocontrol [59]. The epoxide underwent regioselective opening [60,61] leading to the β-hydroxyketone that was temporarily masked as a TES ether.
Reduction of ketone 85 with NaBH4 resulted in the formation of two diastereomeric alcohols in 2:1 ratio. To our delight flash chromatography permitted smooth separation of the two compounds. nOe studies revealed that the main product is 1S-configurated as in most B-seco limonoids suggesting that an equatorial attack of the hydride is slightly preferred over the axial attack. However bearing in mind the elimination issues with β-alkoxy esters during Ireland–Claisen rearrangements discussed above we decided to continue the synthesis with both diastereomers as this elimination is expected to proceed faster in syn-substituted β-alkoxy esters since the hydrogen and the leaving group are in an antiperiplanar arrangement.
Continuing the synthesis with the 1S-isomer (Scheme 12), reduction of the ester moiety could be initiated by LiBH4 but proceeded sluggishly. For the protection of the corresponding diol we were limited to small protecting groups as introduction of the MOM-groups already occurred slowly and required high excess of reagents. Selective deprotection of the TBS and the TES ethers in 86 was achieved under acidic conditions. Oxidation of the resulting diol with Dess–Martin periodinane and subsequent Pinnick oxidation completed the synthesis.
Ireland–Claisen rearrangements with fully decorated A ring. With the suitable substituted fragment 87 in hand, we decided to initially perform the intended rearrangement with one of the model C rings. Thus carboxylic acid 87 was esterified with allylic alcohol 40 to give allyl ester 88 (Scheme 13).
![[1860-5397-10-15-i13]](/bjoc/content/inline/1860-5397-10-15-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: and Attempted Ireland–Claisen rearrangement of allyl ester 88. R1 = MOM, R2 = CO2H.
Scheme 13: and Attempted Ireland–Claisen rearrangement of allyl ester 88. R1 = MOM, R2 = CO2H.
Unfortunately exposure of 88 to the optimized conditions developed for the synthesis of the B-seco limonoid scaffold did not initiate the desired Ireland–Claisen rearrangement. Addition of HMPA was also fruitless. Noteworthy elimination of the MOM-protected hydroxy group at C1 occurred only to a minor extent under these conditions, but was not observed at all when HMPA was added. In situ formation of the TMS enol ether of 88 was detected, however as a consequence of the acidic work-up β-elimination of the OMOM group was induced resulting in α,β-unsaturated ketone 90.
Despite these unsatisfactory results we continued with the synthesis of C1-epi allyl ester 93 starting from alcohol 91 employing the same reaction sequence of ester reduction, diol protection, desilylation, oxidation and esterification (Scheme 14). Attempts to rearrange 93 under the optimized conditions were again unsuccessful. As expected, elimination of the OMOM group during the reaction was not detected due to the unfavoured orientation of the hydrogen and the leaving group. Subsequent acidic treatment did again lead to formation of enone 90.
![[1860-5397-10-15-i14]](/bjoc/content/inline/1860-5397-10-15-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis and attempted Ireland–Claisen rearrangement of allyl ester 93. Reagents and conditions: a) LiBH4, THF, 65 °C, overnight, 84%; b) MOMCl, DIPEA, NaI, THF, 50 °C, overnight, 89%; c) 8% TFA in CH2Cl2, rt, 0.5 h, 52%; d) DMP, NaHCO3, CH2Cl2, 0 °C, 3 h, 86%; e) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O (4:1), rt, 2.5 h, 99%; f) EDC·HCl, 40, DMAP, CH2Cl2, rt, 2 d, 47%. R1 = MOM, R2 = CO2H.
Scheme 14: Synthesis and attempted Ireland–Claisen rearrangement of allyl ester 93. Reagents and conditions: a...
We assume that the additional substituents as well as the intermediary formed TMS ether cause excessive steric bulk and rigidity that prevent the silyl ketene acetals from adopting the required conformation. Hence, we considered enone 90 as an alternative rearrangement precursor as many B-seco limonoids exhibit a double bond in this position (see 21-hydroxytoonacilide (1)). However, despite the incapacity to form the silyl enol ether as well as the lack of the C1-substituent allyl ester 90 failed to furnish the corresponding carboxylic acid using the general conditions.
In order to explore whether the whole system or whether the A ring 87 itself is too crowded to allow the rearrangement to proceed, we used a completely undecorated C ring. For this purpose rearrangement precursors 95, 96, and 97 were synthesized (see Supporting Information File 1) and investigated with respect to their behavior in the [3,3]-sigmatropic rearrangement (Scheme 15). Unfortunately all attempts to procure this transformation using different bases (LiHMDS, KHMDS, LDA), additives (Et3N, HMPA), solvents (THF, toluene) and temperatures (up to 110 °C) completely failed. Indeed the results are in accordance with those obtained earlier. All allyl esters were converted to the corresponding TMS ethers under the rearrangement conditions. Formation of the β-elimination product was exclusively observed for the 1S-configurated allyl ester 95 in the absence of HMPA. Replacing the sterically demanding TBDPS group by the smaller TBS group in 97 could not initiate the rearrangement either. Furthermore, α,β-unsaturated ketone 98, that was generated during the acidic work-up, did not show any conversion (Scheme 15).
![[1860-5397-10-15-i15]](/bjoc/content/inline/1860-5397-10-15-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Allyl esters tested in the Ireland–Claisen rearrangement.
Scheme 15: Allyl esters tested in the Ireland–Claisen rearrangement.
Conclusion
In view of the results obtained we conclude that an Ireland–Claisen rearrangement is not a suitable method to build up the completely decorated scaffold of the B-seco limonoid natural products, as it failed when a fully substituted A ring is used. Presumably steric constraints in combination with the rigidity caused by the intermediary formed silyl enol ether are responsible for the failure as it has been indicated before for substrates that contain a rigid BDA group or a bulky TIPS group.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterisation data. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Mulholland, D. A.; Parel, B.; Coombes, P. H. Curr. Org. Chem. 2000, 4, 1011–1054. doi:10.2174/1385272003375941
Return to citation in text: [1] -
Roy, A.; Saraf, S. Biol. Pharm. Bull. 2006, 29, 191–201. doi:10.1248/bpb.29.191
Return to citation in text: [1] -
Heasley, B. Eur. J. Org. Chem. 2011, 19–46. doi:10.1002/ejoc.201001218
Return to citation in text: [1] -
Kraus, W.; Grimminger, W. Nouv. J. Chim. 1980, 4, 651–655.
Return to citation in text: [1] -
Cheplogoi, P. K.; Mulholland, D. A. Phytochemistry 2003, 62, 1173–1178. doi:10.1016/S0031-9422(03)00028-1
Return to citation in text: [1] -
Lidert, Z.; Taylor, D. A. H.; Thirugnanam, M. J. Nat. Prod. 1985, 48, 843–845. doi:10.1021/np50041a029
Return to citation in text: [1] [2] -
Sarker, S. D.; Savchenko, T.; Whiting, P.; Šik, V.; Dinan, L. Arch. Insect Biochem. Physiol. 1997, 35, 211–217. doi:10.1002/(SICI)1520-6327(1997)35:1/2<211::AID-ARCH19>3.0.CO;2-E
Return to citation in text: [1] [2] -
Koul, O.; Daniewski, W. M.; Multani, J. S.; Gumulka, M.; Singh, G. J. Agric. Food Chem. 2003, 51, 7271–7275. doi:10.1021/jf0304223
Return to citation in text: [1] -
Bray, D. H.; Warhurst, D. C.; Connolly, J. D.; O'Neill, M. J.; Phillipson, J. D. Phytother. Res. 1990, 4, 29–35. doi:10.1002/ptr.2650040108
Return to citation in text: [1] -
Lukacova, V.; Polonsky, J.; Moretti, C.; Pettit, G. R.; Schmidt, J. M. J. Nat. Prod. 1982, 45, 288–294. doi:10.1021/np50021a010
Return to citation in text: [1] [2] -
Pettit, G. R.; Barton, D. H. R.; Herald, C. L.; Polonsky, J.; Schmidt, J. M.; Connolly, J. D. J. Nat. Prod. 1983, 46, 379–390. doi:10.1021/np50027a015
Return to citation in text: [1] [2] -
Musza, L. L.; Killar, L. M.; Speight, P.; McElhiney, S.; Barrow, C. J.; Gillum, A. M.; Cooper, R. Tetrahedron 1994, 50, 11369–11378. doi:10.1016/S0040-4020(01)89279-6
Return to citation in text: [1] -
Bevan, C. W. L.; Ekong, D. E. U.; Taylor, D. A. H. Nature 1965, 206, 1323–1325. doi:10.1038/2061323a0
Return to citation in text: [1] -
Gullo, V. P.; Miura, I.; Nakanishi, K.; Cameron, A. F.; Connolly, J. D.; Duncanson, F. D.; Harding, A. E.; McCrindle, R.; Taylor, D. A. H. J. Chem. Soc., Chem. Commun. 1975, 345–346. doi:10.1039/c39750000345
Return to citation in text: [1] -
Cameron, A. F.; Duncanson, F. D. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1976, 32, 1841–1845. doi:10.1107/S056774087600650X
Return to citation in text: [1] -
Robert, S.; Chary, S. N.; Drakakaki, G.; Li, S.; Yang, Z.; Raikhel, N. V.; Hicks, G. R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8464–8469. doi:10.1073/pnas.0711650105
Return to citation in text: [1] -
Kraus, W.; Grimminger, W.; Sawitzki, G. Angew. Chem., Int. Ed. Engl. 1978, 17, 452–453. doi:10.1002/anie.197804521
Return to citation in text: [1] -
Munehiro, N.; Miyako, O.; Takashi, I.; Kosei, M.; Hideo, N.; Tsunao, H. Heterocycles 1984, 22, 2335–2340. doi:10.3987/R-1984-10-2335
Return to citation in text: [1] -
Luo, X.-D.; Wu, S.-H.; Wu, D.-G.; Ma, Y.-B.; Qi, S.-H. Tetrahedron 2002, 58, 7797–7804. doi:10.1016/S0040-4020(02)00944-4
Return to citation in text: [1] -
Koul, O.; Daniewski, W. M.; Multani, J. S.; Gumulka, M.; Singh, G. J. Agric. Food Chem. 2003, 51, 7271–7275. doi:10.1021/jf0304223
Return to citation in text: [1] -
Qi, S.-H.; Chen, L.; Wu, D.-G.; Ma, Y.-B.; Luo, X.-D. Tetrahedron 2003, 59, 4193–4199. doi:10.1016/S0040-4020(03)00573-8
Return to citation in text: [1] -
Wang, X.-N.; Yin, S.; Fan, C.-Q.; Wang, F.-D.; Lin, L.-P.; Ding, J.; Yue, J.-M. Org. Lett. 2006, 8, 3845–3848. doi:10.1021/ol061466a
Return to citation in text: [1] -
Wang, X.-N.; Yin, S.; Fan, C.-Q.; Lin, L.-P.; Ding, J.; Yue, J.-M. Tetrahedron 2007, 63, 8234–8241. doi:10.1016/j.tet.2007.05.107
Return to citation in text: [1] -
Kishore, K. C.; Vijayalakshmi, K.; Bibha, C.; Mridula, N.; Rama Gopal, G.; Sathees, C. R. FEBS Lett. 2008, 582, 4066–4076. doi:10.1016/j.febslet.2008.11.001
Return to citation in text: [1] -
Yang, S.-P.; Chen, H.-D.; Liao, S.-G.; Xie, B.-J.; Miao, Z.-H.; Yue, J.-M. Org. Lett. 2011, 13, 150–153. doi:10.1021/ol102745h
Return to citation in text: [1] -
Tóth, R.; Gerding-Reimers, C.; Deeks, M. J.; Menninger, S.; Gallegos, R. M.; Tonaco, I. A. N.; Hübel, K.; Hussey, P. J.; Waldmann, H.; Coupland, G. Plant J. 2012, 71, 338–352. doi:10.1111/j.1365-313X.2012.04991.x
Return to citation in text: [1] -
Wetzel, S.; Bon, R. S.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2011, 50, 10800–10826. doi:10.1002/anie.201007004
Return to citation in text: [1] -
Bon, R. S.; Waldmann, H. Acc. Chem. Res. 2010, 43, 1103–1114. doi:10.1021/ar100014h
Return to citation in text: [1] -
Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2009, 48, 3224–3242. doi:10.1002/anie.200803437
Return to citation in text: [1] -
Nören-Müller, A.; Reis-Correa, I., Jr.; Prinz, H.; Rosenbaum, C.; Saxena, K.; Schwalbe, H. J.; Vestweber, D.; Cagna, G.; Schunk, S.; Schwarz, O.; Schiewe, H.; Waldmann, H. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10606–10611. doi:10.1073/pnas.0601490103
Return to citation in text: [1] -
Koch, M. A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17272–17277. doi:10.1073/pnas.0503647102
Return to citation in text: [1] -
Koch, M. A.; Wittenberg, L.-O.; Basu, S.; Jeyaraj, D. A.; Gourzoulidou, E.; Reinecke, K.; Odermatt, A.; Waldmann, H. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16721–16726. doi:10.1073/pnas.0404719101
Return to citation in text: [1] -
Dückert, H.; Pries, V.; Khedkar, V.; Menninger, S.; Bruss, H.; Bird, A. W.; Maliga, Z.; Brockmeyer, A.; Janning, P.; Hyman, A.; Grimme, S.; Schürmann, M.; Preut, H.; Hübel, K.; Ziegler, S.; Kumar, K.; Waldmann, H. Nat. Chem. Biol. 2012, 8, 179–184. doi:10.1038/nchembio.758
Return to citation in text: [1] -
Zimmermann, T. J.; Roy, S.; Martinez, N. E.; Ziegler, S.; Hedberg, C.; Waldmann, H. ChemBioChem 2013, 14, 295–300. doi:10.1002/cbic.201200711
Return to citation in text: [1] -
In all molecules described in this paper including fragments and model compounds we maintain the numbering of the carbon atoms established for limonoid natural products as it is indicated in Figure 1 for 21-hydroxytoonacilide (1).
Return to citation in text: [1] -
Schuster, H.; Martinez, R.; Bruss, H.; Antonchick, A. P.; Kaiser, M.; Schürmann, M.; Waldmann, H. Chem. Commun. 2011, 47, 6545–6547. doi:10.1039/c1cc11388g
Return to citation in text: [1] [2] -
Veitch, G. E.; Beckmann, E.; Burke, B. J.; Boyer, A.; Maslen, S. L.; Ley, S. V. Angew. Chem., Int. Ed. 2007, 46, 7629–7632. doi:10.1002/anie.200703027
Return to citation in text: [1] -
Veitch, G. E.; Beckmann, E.; Burke, B. J.; Boyer, A.; Ayats, C.; Ley, S. V. Angew. Chem., Int. Ed. 2007, 46, 7633–7635. doi:10.1002/anie.200703028
Return to citation in text: [1] -
Barros, M. T.; Maycock, C. D.; Ventura, M. R. J. Chem. Soc., Perkin Trans. 1 2001, 166–173. doi:10.1039/b002980g
Return to citation in text: [1] [2] -
Arthurs, C. L.; Lingley, K. F.; Piacenti, M.; Stratford, I. J.; Tatic, T.; Whitehead, R. C.; Wind, N. S. Tetrahedron Lett. 2008, 49, 2410–2413. doi:10.1016/j.tetlet.2008.02.059
Return to citation in text: [1] -
Murray, L. M.; O'Brien, P.; Taylor, R. J. K. Org. Lett. 2003, 5, 1943–1946. doi:10.1021/ol034521d
Return to citation in text: [1] [2] -
Marchart, S.; Mulzer, J.; Enev, V. S. Org. Lett. 2007, 9, 813–816. doi:10.1021/ol0630189
Return to citation in text: [1] -
Lence, E.; Castedo, L.; Gonzalez-Bello, C. Chem. Soc. Rev. 2008, 37, 1689–1708. doi:10.1039/b717902b
Return to citation in text: [1] -
Durand-Reville, T.; Gobbi, L. B.; Gray, B. L.; Ley, S. V.; Scott, J. S. Org. Lett. 2002, 4, 3847–3850. doi:10.1021/ol0201557
Return to citation in text: [1] -
Ziegler, F. E.; Nangia, A.; Schulte, G. Tetrahedron Lett. 1988, 29, 1669–1672. doi:10.1016/S0040-4039(00)82013-4
Return to citation in text: [1] -
Quach, T. D.; Batey, R. A. Org. Lett. 2003, 5, 1381–1384. doi:10.1021/ol034454n
Return to citation in text: [1] -
Nordmann, G.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 4978–4979. doi:10.1021/ja034809y
Return to citation in text: [1] -
Fukuzaki, T.; Kobayashi, S.; Hibi, T.; Ikuma, Y.; Ishihara, J.; Kanoh, N.; Murai, A. Org. Lett. 2002, 4, 2877–2880. doi:10.1021/ol026260y
Return to citation in text: [1] -
Ireland, R. E.; Varney, M. D. J. Org. Chem. 1983, 48, 1829–1833. doi:10.1021/jo00159a008
Return to citation in text: [1] -
Hiersemann, M.; Nubbemeyer, U., Eds. The Claisen Rearrangement; Wiley-VCH: Weinheim, Germany, 2007.
Return to citation in text: [1] -
Chieffi, A.; Kamikawa, K.; Åhman, J.; Fox, J. M.; Buchwald, S. L. Org. Lett. 2001, 3, 1897–1900. doi:10.1021/ol0159470
Return to citation in text: [1] -
Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615–1621. doi:10.1021/jo00925a003
Return to citation in text: [1] -
Arseniyadis, S.; Yashunsky, D. V.; de Freitas, R. P.; Dorado, M. M.; Potier, P.; Toupet, L. Tetrahedron 1996, 52, 12443–12458. doi:10.1016/0040-4020(96)00735-1
Return to citation in text: [1] -
Hudson, P.; Parsons, P. J. Synlett 1992, 867–868. doi:10.1055/s-1992-21520
Return to citation in text: [1] -
Shi, H. Synth. Commun. 2006, 36, 237–248. doi:10.1080/00397910500334611
Return to citation in text: [1] -
Ito, Y.; Hirao, T.; Saegusa, T. J. Org. Chem. 1978, 43, 1011–1013. doi:10.1021/jo00399a052
Return to citation in text: [1] -
Larock, R. C.; Hightower, T. R.; Kraus, G. A.; Hahn, P.; Zheng, D. Tetrahedron Lett. 1995, 36, 2423–2426. doi:10.1016/0040-4039(95)00306-W
Return to citation in text: [1] -
Deguest, G.; Bischoff, L.; Fruit, C.; Marsais, F. Org. Lett. 2007, 9, 1165–1167. doi:10.1021/ol070145b
Return to citation in text: [1] -
The configuration of the epoxide was determined via Mosher ester analysis of a derivative of the corresponding β-hydroxyketone, that was obtained during the synthetic studies.
Return to citation in text: [1] -
Miyashita, M.; Suzuki, T.; Yoshikoshi, A. Tetrahedron Lett. 1987, 28, 4293–4296. doi:10.1016/S0040-4039(00)96488-8
Return to citation in text: [1] -
Miyashita, M.; Suzuki, T.; Hoshino, M.; Yoshikoshi, A. Tetrahedron 1997, 53, 12469–12486. doi:10.1016/S0040-4020(97)00781-3
Return to citation in text: [1]
| 56. | Ito, Y.; Hirao, T.; Saegusa, T. J. Org. Chem. 1978, 43, 1011–1013. doi:10.1021/jo00399a052 |
| 57. | Larock, R. C.; Hightower, T. R.; Kraus, G. A.; Hahn, P.; Zheng, D. Tetrahedron Lett. 1995, 36, 2423–2426. doi:10.1016/0040-4039(95)00306-W |
| 58. | Deguest, G.; Bischoff, L.; Fruit, C.; Marsais, F. Org. Lett. 2007, 9, 1165–1167. doi:10.1021/ol070145b |
| 59. | The configuration of the epoxide was determined via Mosher ester analysis of a derivative of the corresponding β-hydroxyketone, that was obtained during the synthetic studies. |
| 1. | Mulholland, D. A.; Parel, B.; Coombes, P. H. Curr. Org. Chem. 2000, 4, 1011–1054. doi:10.2174/1385272003375941 |
| 2. | Roy, A.; Saraf, S. Biol. Pharm. Bull. 2006, 29, 191–201. doi:10.1248/bpb.29.191 |
| 3. | Heasley, B. Eur. J. Org. Chem. 2011, 19–46. doi:10.1002/ejoc.201001218 |
| 10. | Lukacova, V.; Polonsky, J.; Moretti, C.; Pettit, G. R.; Schmidt, J. M. J. Nat. Prod. 1982, 45, 288–294. doi:10.1021/np50021a010 |
| 11. | Pettit, G. R.; Barton, D. H. R.; Herald, C. L.; Polonsky, J.; Schmidt, J. M.; Connolly, J. D. J. Nat. Prod. 1983, 46, 379–390. doi:10.1021/np50027a015 |
| 22. | Wang, X.-N.; Yin, S.; Fan, C.-Q.; Wang, F.-D.; Lin, L.-P.; Ding, J.; Yue, J.-M. Org. Lett. 2006, 8, 3845–3848. doi:10.1021/ol061466a |
| 23. | Wang, X.-N.; Yin, S.; Fan, C.-Q.; Lin, L.-P.; Ding, J.; Yue, J.-M. Tetrahedron 2007, 63, 8234–8241. doi:10.1016/j.tet.2007.05.107 |
| 24. | Kishore, K. C.; Vijayalakshmi, K.; Bibha, C.; Mridula, N.; Rama Gopal, G.; Sathees, C. R. FEBS Lett. 2008, 582, 4066–4076. doi:10.1016/j.febslet.2008.11.001 |
| 25. | Yang, S.-P.; Chen, H.-D.; Liao, S.-G.; Xie, B.-J.; Miao, Z.-H.; Yue, J.-M. Org. Lett. 2011, 13, 150–153. doi:10.1021/ol102745h |
| 43. | Lence, E.; Castedo, L.; Gonzalez-Bello, C. Chem. Soc. Rev. 2008, 37, 1689–1708. doi:10.1039/b717902b |
| 6. | Lidert, Z.; Taylor, D. A. H.; Thirugnanam, M. J. Nat. Prod. 1985, 48, 843–845. doi:10.1021/np50041a029 |
| 7. | Sarker, S. D.; Savchenko, T.; Whiting, P.; Šik, V.; Dinan, L. Arch. Insect Biochem. Physiol. 1997, 35, 211–217. doi:10.1002/(SICI)1520-6327(1997)35:1/2<211::AID-ARCH19>3.0.CO;2-E |
| 17. | Kraus, W.; Grimminger, W.; Sawitzki, G. Angew. Chem., Int. Ed. Engl. 1978, 17, 452–453. doi:10.1002/anie.197804521 |
| 18. | Munehiro, N.; Miyako, O.; Takashi, I.; Kosei, M.; Hideo, N.; Tsunao, H. Heterocycles 1984, 22, 2335–2340. doi:10.3987/R-1984-10-2335 |
| 19. | Luo, X.-D.; Wu, S.-H.; Wu, D.-G.; Ma, Y.-B.; Qi, S.-H. Tetrahedron 2002, 58, 7797–7804. doi:10.1016/S0040-4020(02)00944-4 |
| 20. | Koul, O.; Daniewski, W. M.; Multani, J. S.; Gumulka, M.; Singh, G. J. Agric. Food Chem. 2003, 51, 7271–7275. doi:10.1021/jf0304223 |
| 21. | Qi, S.-H.; Chen, L.; Wu, D.-G.; Ma, Y.-B.; Luo, X.-D. Tetrahedron 2003, 59, 4193–4199. doi:10.1016/S0040-4020(03)00573-8 |
| 44. | Durand-Reville, T.; Gobbi, L. B.; Gray, B. L.; Ley, S. V.; Scott, J. S. Org. Lett. 2002, 4, 3847–3850. doi:10.1021/ol0201557 |
| 45. | Ziegler, F. E.; Nangia, A.; Schulte, G. Tetrahedron Lett. 1988, 29, 1669–1672. doi:10.1016/S0040-4039(00)82013-4 |
| 6. | Lidert, Z.; Taylor, D. A. H.; Thirugnanam, M. J. Nat. Prod. 1985, 48, 843–845. doi:10.1021/np50041a029 |
| 7. | Sarker, S. D.; Savchenko, T.; Whiting, P.; Šik, V.; Dinan, L. Arch. Insect Biochem. Physiol. 1997, 35, 211–217. doi:10.1002/(SICI)1520-6327(1997)35:1/2<211::AID-ARCH19>3.0.CO;2-E |
| 8. | Koul, O.; Daniewski, W. M.; Multani, J. S.; Gumulka, M.; Singh, G. J. Agric. Food Chem. 2003, 51, 7271–7275. doi:10.1021/jf0304223 |
| 9. | Bray, D. H.; Warhurst, D. C.; Connolly, J. D.; O'Neill, M. J.; Phillipson, J. D. Phytother. Res. 1990, 4, 29–35. doi:10.1002/ptr.2650040108 |
| 10. | Lukacova, V.; Polonsky, J.; Moretti, C.; Pettit, G. R.; Schmidt, J. M. J. Nat. Prod. 1982, 45, 288–294. doi:10.1021/np50021a010 |
| 11. | Pettit, G. R.; Barton, D. H. R.; Herald, C. L.; Polonsky, J.; Schmidt, J. M.; Connolly, J. D. J. Nat. Prod. 1983, 46, 379–390. doi:10.1021/np50027a015 |
| 12. | Musza, L. L.; Killar, L. M.; Speight, P.; McElhiney, S.; Barrow, C. J.; Gillum, A. M.; Cooper, R. Tetrahedron 1994, 50, 11369–11378. doi:10.1016/S0040-4020(01)89279-6 |
| 13. | Bevan, C. W. L.; Ekong, D. E. U.; Taylor, D. A. H. Nature 1965, 206, 1323–1325. doi:10.1038/2061323a0 |
| 14. | Gullo, V. P.; Miura, I.; Nakanishi, K.; Cameron, A. F.; Connolly, J. D.; Duncanson, F. D.; Harding, A. E.; McCrindle, R.; Taylor, D. A. H. J. Chem. Soc., Chem. Commun. 1975, 345–346. doi:10.1039/c39750000345 |
| 15. | Cameron, A. F.; Duncanson, F. D. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1976, 32, 1841–1845. doi:10.1107/S056774087600650X |
| 16. | Robert, S.; Chary, S. N.; Drakakaki, G.; Li, S.; Yang, Z.; Raikhel, N. V.; Hicks, G. R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8464–8469. doi:10.1073/pnas.0711650105 |
| 40. | Arthurs, C. L.; Lingley, K. F.; Piacenti, M.; Stratford, I. J.; Tatic, T.; Whitehead, R. C.; Wind, N. S. Tetrahedron Lett. 2008, 49, 2410–2413. doi:10.1016/j.tetlet.2008.02.059 |
| 4. | Kraus, W.; Grimminger, W. Nouv. J. Chim. 1980, 4, 651–655. |
| 5. | Cheplogoi, P. K.; Mulholland, D. A. Phytochemistry 2003, 62, 1173–1178. doi:10.1016/S0031-9422(03)00028-1 |
| 41. | Murray, L. M.; O'Brien, P.; Taylor, R. J. K. Org. Lett. 2003, 5, 1943–1946. doi:10.1021/ol034521d |
| 42. | Marchart, S.; Mulzer, J.; Enev, V. S. Org. Lett. 2007, 9, 813–816. doi:10.1021/ol0630189 |
| 36. | Schuster, H.; Martinez, R.; Bruss, H.; Antonchick, A. P.; Kaiser, M.; Schürmann, M.; Waldmann, H. Chem. Commun. 2011, 47, 6545–6547. doi:10.1039/c1cc11388g |
| 39. | Barros, M. T.; Maycock, C. D.; Ventura, M. R. J. Chem. Soc., Perkin Trans. 1 2001, 166–173. doi:10.1039/b002980g |
| 35. | In all molecules described in this paper including fragments and model compounds we maintain the numbering of the carbon atoms established for limonoid natural products as it is indicated in Figure 1 for 21-hydroxytoonacilide (1). |
| 39. | Barros, M. T.; Maycock, C. D.; Ventura, M. R. J. Chem. Soc., Perkin Trans. 1 2001, 166–173. doi:10.1039/b002980g |
| 27. | Wetzel, S.; Bon, R. S.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2011, 50, 10800–10826. doi:10.1002/anie.201007004 |
| 28. | Bon, R. S.; Waldmann, H. Acc. Chem. Res. 2010, 43, 1103–1114. doi:10.1021/ar100014h |
| 29. | Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2009, 48, 3224–3242. doi:10.1002/anie.200803437 |
| 30. | Nören-Müller, A.; Reis-Correa, I., Jr.; Prinz, H.; Rosenbaum, C.; Saxena, K.; Schwalbe, H. J.; Vestweber, D.; Cagna, G.; Schunk, S.; Schwarz, O.; Schiewe, H.; Waldmann, H. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10606–10611. doi:10.1073/pnas.0601490103 |
| 31. | Koch, M. A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17272–17277. doi:10.1073/pnas.0503647102 |
| 32. | Koch, M. A.; Wittenberg, L.-O.; Basu, S.; Jeyaraj, D. A.; Gourzoulidou, E.; Reinecke, K.; Odermatt, A.; Waldmann, H. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16721–16726. doi:10.1073/pnas.0404719101 |
| 33. | Dückert, H.; Pries, V.; Khedkar, V.; Menninger, S.; Bruss, H.; Bird, A. W.; Maliga, Z.; Brockmeyer, A.; Janning, P.; Hyman, A.; Grimme, S.; Schürmann, M.; Preut, H.; Hübel, K.; Ziegler, S.; Kumar, K.; Waldmann, H. Nat. Chem. Biol. 2012, 8, 179–184. doi:10.1038/nchembio.758 |
| 34. | Zimmermann, T. J.; Roy, S.; Martinez, N. E.; Ziegler, S.; Hedberg, C.; Waldmann, H. ChemBioChem 2013, 14, 295–300. doi:10.1002/cbic.201200711 |
| 60. | Miyashita, M.; Suzuki, T.; Yoshikoshi, A. Tetrahedron Lett. 1987, 28, 4293–4296. doi:10.1016/S0040-4039(00)96488-8 |
| 61. | Miyashita, M.; Suzuki, T.; Hoshino, M.; Yoshikoshi, A. Tetrahedron 1997, 53, 12469–12486. doi:10.1016/S0040-4020(97)00781-3 |
| 26. | Tóth, R.; Gerding-Reimers, C.; Deeks, M. J.; Menninger, S.; Gallegos, R. M.; Tonaco, I. A. N.; Hübel, K.; Hussey, P. J.; Waldmann, H.; Coupland, G. Plant J. 2012, 71, 338–352. doi:10.1111/j.1365-313X.2012.04991.x |
| 37. | Veitch, G. E.; Beckmann, E.; Burke, B. J.; Boyer, A.; Maslen, S. L.; Ley, S. V. Angew. Chem., Int. Ed. 2007, 46, 7629–7632. doi:10.1002/anie.200703027 |
| 38. | Veitch, G. E.; Beckmann, E.; Burke, B. J.; Boyer, A.; Ayats, C.; Ley, S. V. Angew. Chem., Int. Ed. 2007, 46, 7633–7635. doi:10.1002/anie.200703028 |
| 48. | Fukuzaki, T.; Kobayashi, S.; Hibi, T.; Ikuma, Y.; Ishihara, J.; Kanoh, N.; Murai, A. Org. Lett. 2002, 4, 2877–2880. doi:10.1021/ol026260y |
| 46. | Quach, T. D.; Batey, R. A. Org. Lett. 2003, 5, 1381–1384. doi:10.1021/ol034454n |
| 47. | Nordmann, G.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 4978–4979. doi:10.1021/ja034809y |
| 53. | Arseniyadis, S.; Yashunsky, D. V.; de Freitas, R. P.; Dorado, M. M.; Potier, P.; Toupet, L. Tetrahedron 1996, 52, 12443–12458. doi:10.1016/0040-4020(96)00735-1 |
| 54. | Hudson, P.; Parsons, P. J. Synlett 1992, 867–868. doi:10.1055/s-1992-21520 |
| 55. | Shi, H. Synth. Commun. 2006, 36, 237–248. doi:10.1080/00397910500334611 |
| 36. | Schuster, H.; Martinez, R.; Bruss, H.; Antonchick, A. P.; Kaiser, M.; Schürmann, M.; Waldmann, H. Chem. Commun. 2011, 47, 6545–6547. doi:10.1039/c1cc11388g |
| 52. | Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615–1621. doi:10.1021/jo00925a003 |
| 51. | Chieffi, A.; Kamikawa, K.; Åhman, J.; Fox, J. M.; Buchwald, S. L. Org. Lett. 2001, 3, 1897–1900. doi:10.1021/ol0159470 |
| 41. | Murray, L. M.; O'Brien, P.; Taylor, R. J. K. Org. Lett. 2003, 5, 1943–1946. doi:10.1021/ol034521d |
| 49. | Ireland, R. E.; Varney, M. D. J. Org. Chem. 1983, 48, 1829–1833. doi:10.1021/jo00159a008 |
| 50. | Hiersemann, M.; Nubbemeyer, U., Eds. The Claisen Rearrangement; Wiley-VCH: Weinheim, Germany, 2007. |
© 2014 Bruss et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)