Abstract
Background
Prior work from these laboratories has centred on the development of enaminones as versatile intermediates for the synthesis of alkaloids and other nitrogen-containing heterocycles. In this paper we describe the enantioselective synthesis of indolizidine and quinolizidine analogues of bicyclic amphibian alkaloids via pyrrolidinylidene- and piperidinylidene-containing enaminones.
Results
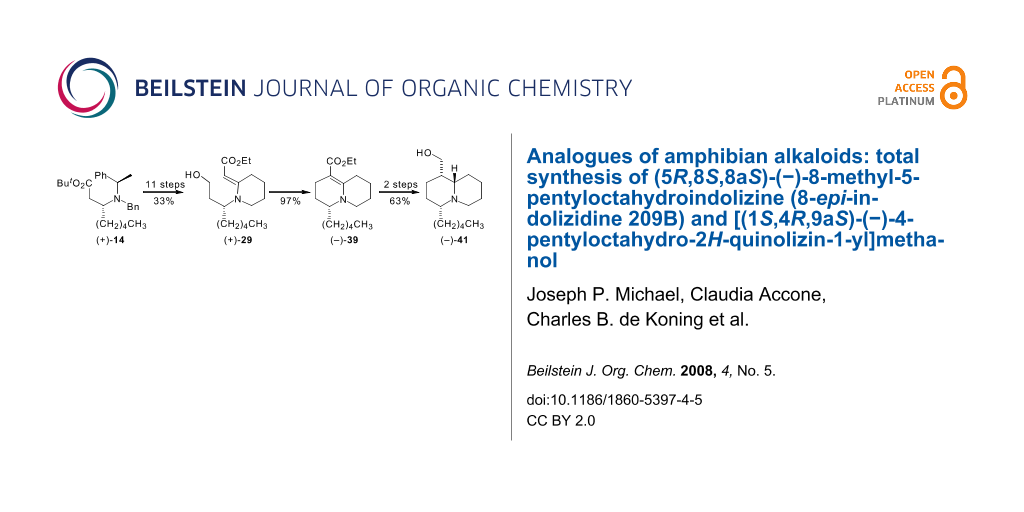
Our previously reported synthesis of racemic 8-epi-indolizidine 209B has been extended to the laevorotatory enantiomer, (−)-9. Attempts to adapt the synthetic route in order to obtain quinolizidine analogues revealed that a key piperidinylidene-containing enaminone intermediate (+)-28 was less tractable than its pyrrolidinylidene counterpart, thereby necessitating modifications that included timing changes and additional protection–deprotection steps. A successful synthesis of [(1S,4R,9aS)-4-pentyloctahydro-2H-quinolizin-1-yl]methanol (−)-41 from the chiral amine tert-butyl (3R)-3-{benzyl[(1R)-1-phenylethyl]amino}octanoate (+)-14 was achieved in 14 steps and an overall yield of 20.4%.
Conclusion
The methodology reported in this article was successfully applied to the enantioselective synthesis of the title compounds. It paves the way for the total synthesis of a range of cis-5,8-disubstituted indolizidines and cis-1,4-disubstituted quinolizidines, as well as the naturally occurring trans-disubstituted alkaloids.

Graphical Abstract
Background
The astonishingly diverse range of alkaloids isolated from the skins of amphibians includes numerous 1-azabicyclic systems belonging to the indolizidine (1-azabicyclo[4.3.0]nonane), quinolizidine (1-azabicyclo[4.4.0]decane) and lehmizidine (1-azabicyclo[5.3.0]decane) classes [1,2]. The first of these classes is by far the most populous, and has commanded enormous attention from organic chemists stimulated by the challenges of designing novel total syntheses [3]. The more recently discovered amphibian quinolizidines constitute a smaller group of alkaloids; they embrace homopumiliotoxins (e.g. (+)-homopumiliotoxin 223G 1; Figure 1) and related systems, 4,6-disubstituted quinolizidines (e.g. rel-quinolizidine 195C 2) and 1,4-disubstituted quinolizidines (e.g. (−)-quinolizidine 217A 3). In the latter group, it appears that most of the well-characterised alkaloids have a 1,4-trans disposition of the substituents; the only alkaloid in which the substituents are unambiguously cis is (−)-quinolizidine 207I 4. Comparatively few syntheses of quinolizidine 207I, 217A and related compounds have been reported [4-9].
![[1860-5397-4-5-1]](/bjoc/content/figures/1860-5397-4-5-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative quinolizidine alkaloids from amphibians.
Figure 1: Representative quinolizidine alkaloids from amphibians.
As part of a long-standing investigation into the utility of pyrrolidinylidene- and piperidinylidene-containing enaminones (vinylogous urethanes) 5 and 6 as key intermediates in the synthesis of alkaloids and other nitrogen-containing heterocycles [10], we previously reported total syntheses of (−)-indolizidine 167B 7 [11,12], the 5,8-disubstituted indolizidine (−)-209B 8 and its racemic diastereomer (±)-9 [13], and the 5,6,8-trisubstituted indolizidines (+)-10 and (+)-11 [14], among other similar compounds (Figure 2). While our attempts to prepare quinolizidines have been less successful, we have synthesised two simple lupin alkaloids, lupinine 12 and epilupinine 13, in racemic form [15]. Although it might seem that reactions of the enaminones 5 and 6 should be directly comparable, we [15,16] and others [17,18] have previously found unexpected differences in the preparation and reactions of cyclic enaminones of different ring sizes. In this article we report our progress in preparing 1,4-disubstituted quinolizidine analogues of amphibian alkaloids by an extension of our approach to the synthesis of 5,8-disubstituted indolizidine alkaloids [19].
![[1860-5397-4-5-2]](/bjoc/content/figures/1860-5397-4-5-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Indolizidines and quinolizidines prepared from enaminone precursors 5 and 6. The conventional numbering scheme for both bicyclic systems is also shown.
Figure 2: Indolizidines and quinolizidines prepared from enaminone precursors 5 and 6. The conventional numbe...
Results and Discussion
Steps in our reported total synthesis of (−)-indolizidine (−)-209B 8 [13] are shown in Scheme 1. Absolute stereocontrol resulted from use of the Davies protocol [20,21], whereby the homochiral amine (+)-14 prepared from tert-butyl (E)-oct-2-enoate and (R)-N-benzyl-1-phenylethylamine, was converted into the primary amine (−)-15 and thence in several steps into the thiolactam (+)-16. Eschenmoser sulfide contraction [22,23] with ethyl bromoacetate yielded the key enaminone intermediate (+)-17, chemoselective reduction of the saturated ester of which produced the alcohol (−)-18. The bicyclic core of the alkaloid was then constructed by a cycloalkylation that took advantage of the nucleophilic reactivity of the enaminone, following which a chemoselective and reasonably diastereoselective (88:12) reduction of the alkene bond of the bicyclic enaminone (+)-19 set up the desired stereochemistry at C-8 and C-8a. Epimerisation of the ester in the reduced compound (−)-20 produced (−)-21, reduction of which gave the alcohol (−)-22. Reduction of the corresponding methanesulfonate with lithium triethylborohydride, as described by Holmes et al. [24], completed the total synthesis of (−)-indolizidine 209B 8.
![[1860-5397-4-5-i1]](/bjoc/content/inline/1860-5397-4-5-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents: (i) H2 (7 atm), 10% Pd/C, AcOH, rt; (ii) Cl(CH2)3COCl, NaHCO3, CHCl3, reflux; (iii) KOBut, ButOH, rt; (iv) Lawesson's reagent, PhMe, reflux; (v) BrCH2CO2Et, MeCN, rt; (vi) Ph3P, Et3N, MeCN, rt; (vii) LiAlH4, THF, rt; (viii) I2, imidazole, Ph3P, PhMe, 110 °C; (ix) H2 (1 atm), PtO2, AcOH, rt; (x) NaOEt (cat.), EtOH, reflux; (xi) LiAlH4, THF, 0 °C to rt; (xii) MeSO2Cl, NEt3, CH2Cl2, 0 °C to rt; (xiii) LiEt3BH, THF, 0 °C.
Scheme 1: Reagents: (i) H2 (7 atm), 10% Pd/C, AcOH, rt; (ii) Cl(CH2)3COCl, NaHCO3, CHCl3, reflux; (iii) KOBut...
As a postscript to the above synthesis, we have now completed an enantioselective synthesis (Scheme 2) of the indolizidine analogue of 8, viz. (5R,8S,8aS)-8-epi-indolizidine 209B (−)-9, which we had previously made as a racemate [13]. Intermediate (−)-20 was reduced with lithium aluminium hydride in diethyl ether to give the alcohol (−)-23 in 97% yield (See Supporting Information File 1 for full experimental data). The corresponding methanesulfonate (−)-24 (66%) was then defunctionalised by an improved procedure, which entailed treatment with freshly prepared Raney nickel [25] in boiling ethanol to give (−)-(5R,8S,8aS)-8-methyl-5-pentyloctahydroindolizine (8-epi-indolizidine 209B) 9 in 74% yield. The spectroscopic data for this product agreed with those reported for the racemate. Support for the cis-relationship of the hydrogen atoms at C-5 and C-8a in all of these compounds was provided by Bohlmann bands [26] at ca. 2790 cm−1 in the FTIR spectra, a feature that also implies a trans-disposition of the lone pair and 8a-H across the ring junction.
![[1860-5397-4-5-i2]](/bjoc/content/inline/1860-5397-4-5-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents: (i) LiAlH4, THF, 0 °C to rt; (ii) MeSO2Cl, NEt3, CH2Cl2, 0 °C to rt; (iii) Raney Ni, EtOH, reflux.
Scheme 2: Reagents: (i) LiAlH4, THF, 0 °C to rt; (ii) MeSO2Cl, NEt3, CH2Cl2, 0 °C to rt; (iii) Raney Ni, EtOH...
Extending the route illustrated in Scheme 1 to the synthesis of quinolizidine analogues required initial acylation of the chiral amine (−)-15, prepared as described in our prior work [13], with 5-bromopentanoyl chloride (obtained in two steps from δ-valerolactone) [27,28]. This afforded tert-butyl (3R)-[(5-bromopentanoyl)amino]octanoate (+)-25 in 98% yield (Scheme 3). However, subsequent cyclisation to the lactam (+)-26 was troublesome, giving at best a yield of 52% when performed with sodium hydride and tetrabutylammonium iodide in N,N-dimethylformamide. An effortless thionation of 26 with Lawesson's reagent in boiling toluene produced the thiolactam (+)-27 in 92% yield. Eschenmoser sulfide contraction was then effected by first treating the thiolactam with ethyl bromoacetate, after which reaction of the resulting S-alkylated intermediate with triethyl phosphite and triethylamine in acetonitrile gave the vinylogous urethane (+)-28 in 75% yield.
![[1860-5397-4-5-i3]](/bjoc/content/inline/1860-5397-4-5-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents: (i) Br(CH2)4COCl, NaHCO3, ClCH2CH2Cl, rt; (ii) NaH, Bu4NI, DMF, rt; (iii) Lawesson's reagent, PhMe, reflux; (iv) BrCH2CO2Et, MeCN, rt; (v) P(OEt)3, Et3N, MeCN, rt; (vi) LiAlH4, THF, rt.
Scheme 3: Reagents: (i) Br(CH2)4COCl, NaHCO3, ClCH2CH2Cl, rt; (ii) NaH, Bu4NI, DMF, rt; (iii) Lawesson's reag...
At this stage, however, our fears of the discrepant behaviour of five- and six-membered enaminones proved to be all too well founded. In the indolizidine series, the robust enaminone 17 survived reduction with lithium aluminium hydride, leaving only the saturated ester to be reduced. With the six-membered analogue 28, the enaminone unit was far more susceptible to reduction, and despite many attempts to modify conditions, over-reduction led to a plethora of basic products that could neither be separated nor properly characterised. Although the desired alcohol (+)-29 containing an intact enaminone system could be isolated on occasion, the best yield obtained was 29% when the reaction was not allowed to go to completion. Thus a change of strategy was required to produce 29, the pivotal intermediate from which the quinolizidine nucleus needs to be constructed.
The reduction of the tert-butyl ester clearly needed to be performed at an early stage of the synthesis before the introduction of other incompatible functional groups (lactam, thiolactam, enaminone). The only feasible option was to go back to the chiral amine (+)-14, reduction of which with lithium aluminium hydride gave the unstable amino alcohol (+)-30 in 97% yield as long as the amine was added slowly to a stirred suspension of the hydride in diethyl ether (Scheme 4). If the order of addition were reversed, the best yield obtained was 48%. The amino alcohol was protected as its tert-butyl(dimethyl)silyl ether (−)-31 (99%) before hydrogenolysis of the benzyl groups over Pearlman's catalyst in glacial acetic acid gave the free amine (−)-32 in quantitative yield. Treatment with 5-bromopentanoyl chloride as described above afforded the unstable bromoamide 33 as an orange oil in 89% yield. In this case, cyclisation of the crude intermediate to the lactam (+)-34 was most successfully effected by adding potassium tert-butoxide to a solution of the bromoamide in dry tetrahydrofuran at room temperature, a yield of 81% being obtained by keeping the reaction time short (25 min). To our dismay, however, the attempted thionation of 34 with Lawesson's reagent under a variety of conditions was uniformly unsuccessful, apparently because the silyl ether failed to survive the reaction conditions.
![[1860-5397-4-5-i4]](/bjoc/content/inline/1860-5397-4-5-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents: (i) LiAlH4, Et2O, 0 °C, then add (+)-2 in Et2O, rt; (ii) TBDMSCl, imidazole, DMF, rt; (iii) H2 (5 atm), 20% Pd(OH)2/C, AcOH, rt; (iv) Br(CH2)4COCl, NaHCO3, ClCH2CH2Cl, rt; (v) t-BuOK, THF, rt, 25 min.
Scheme 4: Reagents: (i) LiAlH4, Et2O, 0 °C, then add (+)-2 in Et2O, rt; (ii) TBDMSCl, imidazole, DMF, rt; (ii...
Inelegant though it was, we were forced at this stage to change protecting groups on the alcohol. Fortunately, the drop in yield was not too serious when desilylation of 34 with aqueous hydrofluoric acid to give the free alcohol (+)-35 was followed by acetylation with acetic anhydride in pyridine (Scheme 5). The lactam (+)-36, obtained in an overall yield of 89%, was then successfully thionated with Lawesson's reagent in boiling toluene to give the thiolactam (+)-37 in 94% yield. Finally, reaction with ethyl bromoacetate followed by treatment with triphenylphosphine and triethylamine in acetonitrile give the vinylogous urethane (+)-38 in 80% yield. Hydrolysis of the acetate with potassium carbonate in methanol then afforded the pivotal alcohol (+)-29 (70%). The scene was now set for cyclisation to the quinolizidine system. Immediate conversion of the unstable free alcohol into the corresponding iodide with iodine, triphenylphosphine and imidazole in a mixture of toluene and acetonitrile [29] and heating the reaction mixture under reflux gave the desired 3,4,6,7,8,9-hexahydro-2H-quinolizine-1-carboxylate (−)-39 in 70% yield.
![[1860-5397-4-5-i5]](/bjoc/content/inline/1860-5397-4-5-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reagents: (i) aq. HF (40%), MeOH, rt; (ii) Ac2O, pyridine, 0 °C to rt; (iii) Lawesson's reagent, PhMe, reflux; (iv) BrCH2CO2Et, MeCN, rt; (v) Ph3P, Et3N, MeCN, rt; (vi) K2CO3, MeOH, rt; (vii) I2, PPh3, imidazole, MeCN-PhMe (2:1), reflux; (viii) H2 (1 atm), PtO2, AcOH, rt; (ix) LiAlH4, THF, 0 °C to rt; (x) MeSO2Cl, NEt3, CH2Cl2, 0 °C to rt; (xi) Raney Ni, EtOH, reflux.
Scheme 5: Reagents: (i) aq. HF (40%), MeOH, rt; (ii) Ac2O, pyridine, 0 °C to rt; (iii) Lawesson's reagent, Ph...
In order to introduce the remaining stereogenic centres of the target system, the alkene bond of the bicyclic vinylogous urethane 39 needs to be reduced stereoselectively. Based on our previous success with the indolizidine analogue 19, we opted for catalytic hydrogenation, which is expected to produce not only a cis-relationship between C-1 and C-9a, but also a cis-relationship between C-4 and C-9a. The developing chair conformation of the six-membered ring in the transition state should result in an equatorial preference for the pentyl side chain, which in turn should bias the approach of the reductant towards the more remote face of the double bond. Gratifyingly, hydrogenation of intermediate 39 over platinum oxide catalyst in ethanol at a pressure of five atmospheres produced the quinolizidine (−)-40 as a single diastereomer in 97% yield. The diastereoselectivity is manifestly better than in the indolizidine case. Support for the cis-relationship of the hydrogen atoms at positions C-4 and C-9a and the trans-ring junction in the product was once again provided by Bohlmann bands in the FTIR spectrum at ca. 2790 cm−1. However, further confirmation of the relative stereochemistry by consideration of the 1H NMR spectrum was not feasible because overlap of signals prevented the extraction of coupling constants for 1-H and 9a-H.
Finally, reduction of the ester to the primary alcohol (−)-41 was accomplished in moderate yield (65%) with lithium aluminium hydride. Again, coupling constants could not be determined for 1-H and 9a-H. In this case, however, there is good precedent for assigning the relative stereochemistry of the hydroxymethyl substituent at C-1 on the basis of 13C chemical shifts. For example, the chemical shift of C-1 in lupinine 12, which possesses an axial hydroxymethyl substituent, is 38.8 ppm; whereas the corresponding chemical shift in epilupinine 13, the equatorial hydroxymethyl epimer, is 43.8 ppm [30]. The chemical shift difference of about 5 ppm between the C-1 equatorial and axial hydroxymethyl epimers appears to be general for quinolizidines [31]. A similar effect has been reported for 8-hydroxymethylindolizidine epimers, for which the chemical shift difference is even larger (ca 10 ppm) [24]. In the present case, the observed chemical shift of 38.4 ppm for 41 is consistent with an axial disposition of the C-1 substituent, and thus with the expected cis-hydrogenation of 39.
While it would have been desirable to conclude this investigation by preparing (1S,4R,9aS)-4-pentyloctahydro-2H-quinolizine 42, the ring homologue of 8-epi-indolizidine 209B, this target eluded us. Attempts to reduce the corresponding methanesulfonate of 41 with Raney nickel in boiling ethanol gave ambiguous results no matter how we modified the reaction conditions.
Conclusion
Few approaches to 1,4-cis-disubstituted quinolizidines and 5,8-cis-disubstituted indolizidines of amphibian origin have been reported in the literature. Because the route we have devised proceeds through bicyclic enaminone intermediates in which the alkene bond is located between the bridgehead position and the adjacent site, we have a convenient and dependable method for introducing the correct relative stereochemistry at these two sites by means of catalytic hydrogenation. However, the differences in behaviour of pyrrolidinylidene- and piperidinylidene-containing enaminones that we have come to expect [15,16] was again apparent, necessitating several protection-deprotection steps that lengthened the route to the quinolizidine system. Nevertheless, our success in preparing the chiral alcohol 41 opens up a route to quinolizidine alkaloids containing C-1 methyl substituents (provided, of course, that we can find a better method for deoxygenation, probably by radical-mediated reaction). In addition, alkyl homologues at C-1 should be accessible; one could, for example, replace the alcohol by a leaving group that can be displaced by organometallic reagents (e.g. cuprates) of appropriate chain length. Substituents at C-4 can also be varied by choosing appropriate analogues of the chiral amine 14, which should also be available in both enantiomeric forms by the Davies procedure [32]. Finally, since the pendent substituents in the indolizidine series can be induced to adopt a trans-orientation by base-catalysed epimerisation of a carbonyl substituent adjacent to the bridgehead position (cf Scheme 1), it should in principle be possible to effect a similar epimerisation in the quinolizidine series, thereby providing a route to most of the known 1,4-disubstituted amphibian quinolizidine alkaloids.
Supporting Information
| Supporting Information File 1: Analogues of amphibian alkaloids - Full experimental details. The Supporting Information File contains detailed experimental procedures and full characterisation data for all new compounds prepared during the synthesis of the two title compounds. | ||
| Format: DOC | Size: 120.5 KB | Download |
References
-
Daly, J. W.; Garraffo, H. M.; Spande, T. F. Alkaloids from amphibian skins. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Pergamon Press: Amsterdam, 1999; Vol. 13, pp 1–161.
Return to citation in text: [1] -
Daly, J. W.; Spande, T. F.; Garraffo, H. M. J. Nat. Prod. 2005, 68, 1556–1575. doi:10.1021/np0580560
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2007, 24, 191–222. doi:10.1039/b509525p
And previous reviews in the series.
Return to citation in text: [1] -
Toyooka, N.; Tanaka, K.; Momose, T.; Daly, J. W.; Garraffo, H. M. Tetrahedron 1997, 53, 9553–9574. doi:10.1016/S0040-4020(97)00641-8
Return to citation in text: [1] -
Pearson, W. H.; Suga, H. J. Org. Chem. 1998, 63, 9910–9918. doi:10.1021/jo981695d
Return to citation in text: [1] -
Michel, P.; Rassat, A.; Daly, J. W.; Spande, T. F. J. Org. Chem. 2000, 65, 8908–8918. doi:10.1021/jo000666b
Return to citation in text: [1] -
Huang, H.; Spande, T. F.; Panek, J. S. J. Am. Chem. Soc. 2003, 125, 626–627. doi:10.1021/ja028937i
Return to citation in text: [1] -
Kinderman, S. S.; de Gelder, R.; van Maarseveen, J. H.; Schoemaker, H. E.; Hiemstra, H.; Rutjes, F. P. J. T. J. Am. Chem. Soc. 2004, 126, 4100–4101. doi:10.1021/ja039919j
Return to citation in text: [1] -
Maloney, K. M.; Danheiser, R. L. Org. Lett. 2005, 7, 3115–3118. doi:10.1021/ol051185n
Return to citation in text: [1] -
Michael, J. P.; de Koning, C. B.; Gravestock, D.; Hosken, G. D.; Howard, A. S.; Jungmann, C. M.; Krause, R. W. M.; Parsons, A. S.; Pelly, S. C.; Stanbury, T. V. Pure Appl. Chem. 1999, 71, 979–988. doi:10.1351/pac199971060979
Return to citation in text: [1] -
Michael, J. P.; Gravestock, D. Eur. J. Org. Chem. 1998, 865–870. doi:10.1002/(SICI)1099-0690(199805)1998:5<865::AID-EJOC865>3.0.CO;2-3
Return to citation in text: [1] -
Michael, J. P.; Gravestock, D. S. Afr. J. Chem. 1998, 51, 146–157.
Return to citation in text: [1] -
Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h
Return to citation in text: [1] [2] [3] [4] -
Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. Org. Biomol. Chem. 2005, 3, 836–847. doi:10.1039/b418062c
Return to citation in text: [1] -
Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77.
Return to citation in text: [1] [2] [3] -
Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. J. Chem. Soc., Perkin Trans. 1 2001, 2055–2062. doi:10.1039/b103560f
Return to citation in text: [1] [2] -
David, O.; Fargeau-Bellassoued, M.-C.; Lhommet, G. Tetrahedron Lett. 2002, 43, 3471–3474. doi:10.1016/S0040-4039(02)00577-4
Return to citation in text: [1] -
Russowsky, D.; da Silveira Neto, B. A. Tetrahedron Lett. 2004, 45, 1437–1440. doi:10.1016/j.tetlet.2003.12.049
Return to citation in text: [1] -
Cai, G.; Zhu, W.; Ma, D. Tetrahedron 2006, 62, 5697–5708. doi:10.1016/j.tet.2006.03.068
And references cited therein for another approach to the synthesis of quinolizidines and related systems via enaminones.
Return to citation in text: [1] -
Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183–186. doi:10.1016/S0957-4166(00)82354-X
Return to citation in text: [1] -
Costello, J. F.; Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1994, 5, 1999–2008. doi:10.1016/S0957-4166(00)86275-8
Return to citation in text: [1] -
Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Helv. Chim. Acta 1971, 54, 710–734. doi:10.1002/hlca.19710540229
Return to citation in text: [1] -
Shiosaki, K. The Eschenmoser coupling reaction. In Comprehensive Organic Synthesis; Trost, B. M., Ed.; Pergamon Press: Oxford, 1991; Vol. 2, pp 865–892.
Return to citation in text: [1] -
Holmes, A. B.; Smith, A. L.; Williams, S. F.; Hughes, L. R.; Lidert, Z.; Swithenbank, C. J. Org. Chem. 1991, 56, 1393–1405. doi:10.1021/jo00004a012
Return to citation in text: [1] [2] -
Covert, L. W.; Adkins, H. J. Am. Chem. Soc. 1932, 54, 4116–4117. doi:10.1021/ja01349a510
Return to citation in text: [1] -
Bohlmann, F. Chem. Ber. 1985, 91, 2157–2167. doi:10.1002/cber.19580911023
Return to citation in text: [1] -
Bagnall, W. H.; Goodings, E. P.; Wilson, C. L. J. Am. Chem. Soc. 1951, 73, 4794–4798. doi:10.1021/ja01154a094
Return to citation in text: [1] -
Collman, J. P.; Groh, S. E. J. Am. Chem. Soc. 1982, 104, 1391–1403. doi:10.1021/ja00369a041
Return to citation in text: [1] -
Garegg, P. J.; Samuelsson, B. J. Chem. Soc., Perkin Trans. 1 1980, 2866–2869. doi:10.1039/P19800002866
Return to citation in text: [1] -
Wenkert, E.; Chauncy, B.; Dave, K. G.; Jeffcoat, A. R.; Schell, F. M.; Schenk, H. P. J. Am. Chem. Soc. 1973, 95, 8427–8436. doi:10.1021/ja00806a038
Return to citation in text: [1] -
Tourwé, D.; van Binst, G. Heterocycles 1978, 9, 507–533.
Return to citation in text: [1] -
Davies, S. G.; Smith, A. D.; Price, P. D. Tetrahedron: Asymmetry 2005, 16, 2833–2891. doi:10.1016/j.tetasy.2005.08.006
Return to citation in text: [1]
| 1. | Daly, J. W.; Garraffo, H. M.; Spande, T. F. Alkaloids from amphibian skins. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Pergamon Press: Amsterdam, 1999; Vol. 13, pp 1–161. |
| 2. | Daly, J. W.; Spande, T. F.; Garraffo, H. M. J. Nat. Prod. 2005, 68, 1556–1575. doi:10.1021/np0580560 |
| 11. | Michael, J. P.; Gravestock, D. Eur. J. Org. Chem. 1998, 865–870. doi:10.1002/(SICI)1099-0690(199805)1998:5<865::AID-EJOC865>3.0.CO;2-3 |
| 12. | Michael, J. P.; Gravestock, D. S. Afr. J. Chem. 1998, 51, 146–157. |
| 24. | Holmes, A. B.; Smith, A. L.; Williams, S. F.; Hughes, L. R.; Lidert, Z.; Swithenbank, C. J. Org. Chem. 1991, 56, 1393–1405. doi:10.1021/jo00004a012 |
| 10. | Michael, J. P.; de Koning, C. B.; Gravestock, D.; Hosken, G. D.; Howard, A. S.; Jungmann, C. M.; Krause, R. W. M.; Parsons, A. S.; Pelly, S. C.; Stanbury, T. V. Pure Appl. Chem. 1999, 71, 979–988. doi:10.1351/pac199971060979 |
| 13. | Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h |
| 4. | Toyooka, N.; Tanaka, K.; Momose, T.; Daly, J. W.; Garraffo, H. M. Tetrahedron 1997, 53, 9553–9574. doi:10.1016/S0040-4020(97)00641-8 |
| 5. | Pearson, W. H.; Suga, H. J. Org. Chem. 1998, 63, 9910–9918. doi:10.1021/jo981695d |
| 6. | Michel, P.; Rassat, A.; Daly, J. W.; Spande, T. F. J. Org. Chem. 2000, 65, 8908–8918. doi:10.1021/jo000666b |
| 7. | Huang, H.; Spande, T. F.; Panek, J. S. J. Am. Chem. Soc. 2003, 125, 626–627. doi:10.1021/ja028937i |
| 8. | Kinderman, S. S.; de Gelder, R.; van Maarseveen, J. H.; Schoemaker, H. E.; Hiemstra, H.; Rutjes, F. P. J. T. J. Am. Chem. Soc. 2004, 126, 4100–4101. doi:10.1021/ja039919j |
| 9. | Maloney, K. M.; Danheiser, R. L. Org. Lett. 2005, 7, 3115–3118. doi:10.1021/ol051185n |
| 20. | Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183–186. doi:10.1016/S0957-4166(00)82354-X |
| 21. | Costello, J. F.; Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1994, 5, 1999–2008. doi:10.1016/S0957-4166(00)86275-8 |
| 3. |
Michael, J. P. Nat. Prod. Rep. 2007, 24, 191–222. doi:10.1039/b509525p
And previous reviews in the series. |
| 22. | Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Helv. Chim. Acta 1971, 54, 710–734. doi:10.1002/hlca.19710540229 |
| 23. | Shiosaki, K. The Eschenmoser coupling reaction. In Comprehensive Organic Synthesis; Trost, B. M., Ed.; Pergamon Press: Oxford, 1991; Vol. 2, pp 865–892. |
| 15. | Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77. |
| 16. | Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. J. Chem. Soc., Perkin Trans. 1 2001, 2055–2062. doi:10.1039/b103560f |
| 19. |
Cai, G.; Zhu, W.; Ma, D. Tetrahedron 2006, 62, 5697–5708. doi:10.1016/j.tet.2006.03.068
And references cited therein for another approach to the synthesis of quinolizidines and related systems via enaminones. |
| 15. | Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77. |
| 13. | Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h |
| 14. | Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. Org. Biomol. Chem. 2005, 3, 836–847. doi:10.1039/b418062c |
| 13. | Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h |
| 17. | David, O.; Fargeau-Bellassoued, M.-C.; Lhommet, G. Tetrahedron Lett. 2002, 43, 3471–3474. doi:10.1016/S0040-4039(02)00577-4 |
| 18. | Russowsky, D.; da Silveira Neto, B. A. Tetrahedron Lett. 2004, 45, 1437–1440. doi:10.1016/j.tetlet.2003.12.049 |
| 13. | Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h |
| 25. | Covert, L. W.; Adkins, H. J. Am. Chem. Soc. 1932, 54, 4116–4117. doi:10.1021/ja01349a510 |
| 32. | Davies, S. G.; Smith, A. D.; Price, P. D. Tetrahedron: Asymmetry 2005, 16, 2833–2891. doi:10.1016/j.tetasy.2005.08.006 |
| 24. | Holmes, A. B.; Smith, A. L.; Williams, S. F.; Hughes, L. R.; Lidert, Z.; Swithenbank, C. J. Org. Chem. 1991, 56, 1393–1405. doi:10.1021/jo00004a012 |
| 15. | Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77. |
| 16. | Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. J. Chem. Soc., Perkin Trans. 1 2001, 2055–2062. doi:10.1039/b103560f |
| 30. | Wenkert, E.; Chauncy, B.; Dave, K. G.; Jeffcoat, A. R.; Schell, F. M.; Schenk, H. P. J. Am. Chem. Soc. 1973, 95, 8427–8436. doi:10.1021/ja00806a038 |
| 27. | Bagnall, W. H.; Goodings, E. P.; Wilson, C. L. J. Am. Chem. Soc. 1951, 73, 4794–4798. doi:10.1021/ja01154a094 |
| 28. | Collman, J. P.; Groh, S. E. J. Am. Chem. Soc. 1982, 104, 1391–1403. doi:10.1021/ja00369a041 |
| 29. | Garegg, P. J.; Samuelsson, B. J. Chem. Soc., Perkin Trans. 1 1980, 2866–2869. doi:10.1039/P19800002866 |
© 2008 Michael et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)