Abstract
A simple, chemoselective transfer hydrogenation of aryl aldehydes with the aid of Amberlite® resin formate (ARF), a stable H-donor, in the presence of catalytic ruthenium trichloride is described. Aromatic aldehydes and 1,2-diketones are reduced efficiently and selectively, while aryl ketones remain unchanged. Several other potentially reducible groups attached to the aromatic moiety are unaffected.

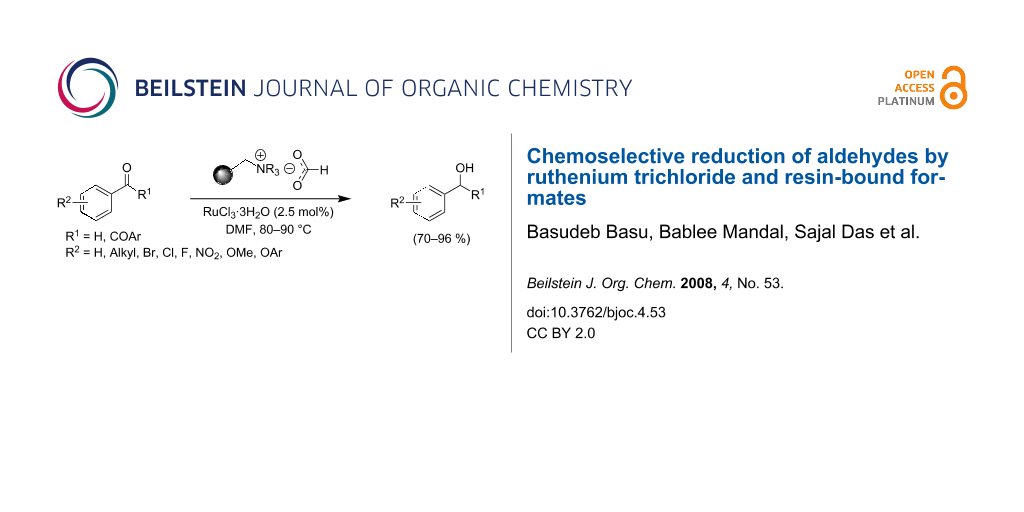
Graphical Abstract
Introduction
Reduction of carbonyl functionality by transition metal-catalyzed transfer hydrogenation (CTH) with the aid of a suitable hydrogen donor is a valuable synthetic tool and has proved to be a viable alternative to hydrogenation using molecular hydrogen [1-3]. Since hydrogenation using molecular hydrogen is associated with risks and often requires high pressure apparatus, the alternative technique, CTH, has been employed in many laboratories. In transfer hydrogenation, several organic molecules such as hydrocarbons [4], primary and secondary alcohols [5,6], and formic acid and its salts [7-11] have been used as the hydrogen source. Besides the use of Rh, Ir, Ni and Pd metals in CTH, carbonyl reduction using the combination of Ru(II)-ligand complexes and propan-2-ol in the presence of a base is a widely used method in modern organic synthesis [5]. The ability of DMA (N,N-dimethylacetamide) or DMF (N,N-dimethylformamide) solutions of RuCl3 to catalyze hydrogenation of simple olefins has long been recognized [12,13]. However, only recently, James and coworkers demonstrated the first example of the use of a simple, phosphine-free, RuCl3-DMA catalytic system in H2-hydrogenation of dimethyl ester of protoporphyrin IX to the mesoporphyrin analogue [14]. Catalytic activity of styrene-divinyl benzene copolymer-bound Ru(III)-EDTA complex was also studied in H2-hydrogenation of alkenes [15]. In the case of CTH, although there are some reports on the use of well-defined ortho-metalated and cyclo-metalated Ru(III) complexes and propan-2-ol (as the hydrogen source) in the presence of a base [16-20], there has been no systematic investigation on the use of RuCl3 in CTH of various organic functional groups.
Reagents immobilized on polymer supports have emerged as potentially attractive tools in terms of clean and green reactions, ease of separation of the products and reusability [21,22]. We have recently demonstrated that poly-ionic resin formate can act as a stable and potent hydride source in Pd-catalyzed transfer hydrogenation of functionalized alkenes, imines, nitroarenes and 1,2-diketones [23,24]. Danks et al. also carried out reduction of alkyl cinnamates using polymer supported formate and catalytic RhCl(PPh3)3 (2.5 mol%) under microwave irradiation [25]. Pd-catalyzed transfer hydrogenation of nitroarenes using recyclable polymer-supported formate has been investigated by Abiraj et al [26]. Neither of these conditions were, however, effective in reducing aryl ketones. Since aryl alcohols are important compounds, we became interested to look at the ability of Ru(III) salts in the CTH of aryl ketones using the poly-ionic resin formate. Our studies reported herein constitute an efficient method for chemoselective transfer hydrogenation of aryl aldehydes with the aid of resin-supported formate in the presence of catalytic (2.5 mol%) amount of commercially available RuCl3·3H2O in DMF or DMA solution (Scheme 1).
![[1860-5397-4-53-i1]](/bjoc/content/inline/1860-5397-4-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: RuCl3-catalyzed transfer hydrogenation of aryl aldehydes.
Scheme 1: RuCl3-catalyzed transfer hydrogenation of aryl aldehydes.
Results and Discussion
In order to optimize the reaction conditions and to find the minimum catalyst requirement, we began our studies with p-anisaldehyde using Amberlite® resin formate (ARF) (0.5 g/mmol of the substrate) as the reducing source. A variety of conditions were investigated, which are summarized in Table 1. Taking 5 mol% of RuCl3·3H2O in DMF and stirring the reaction mixture at 100 °C for 10 h afforded the desired alcohol in 81% yield (Table 1, entry 1). A similar yield of the alcohol was also obtained on carrying out the reaction in presence of 2.5 mol% of RuCl3·3H2O at 80 °C for 8 h (Table 1, entry 6). Further lowering of the amount of RuCl3·3H2O or the reaction temperature, however, led to reduced yield (Table 1, entries 7–9). In order to compare the efficiency of the catalytic combination of the reductant (ARF/RuCl3·3H2O), we carried out the CTH using a well-defined Ru(II) complex [Dichloro(p-cymene)ruthenium(II)] dimer; (2 mol%) under similar conditions and indeed a comparable result was observed (Table 1, entry 11). On the basis of this comparison, it may be presumed that the Ru(III) salt might undergo in situ reduction to Ru(II), which then catalyzes the hydrogenation of the aldehydes.
Table 1: Optimization of the reaction conditions.a
| Entry | Catalyst (mol%) | Temp (°C) | Solvent | t (h) | Yield (%)b |
|---|---|---|---|---|---|
| 1 | 5.0 | 100 | DMF/DMA | 10 | 81 |
| 2 | 5.0 | 80 | DMF | 10 | 84 |
| 3 | 5.0 | 60 | DMF | 10 | 21 |
| 4 | 2.5 | 100 | DMF/DMA | 10 | 80 |
| 5 | 2.5 | 80 | DMF | 10 | 81 |
| 6 | 2.5 | 80 | DMF | 8 | 83 |
| 7 | 1.0 | 80 | DMA | 10 | n.d.c |
| 8 | 2.5 | 60 | DMF | 12 | 29 |
| 9 | 2.0 | 100 | DMA | 12 | 41 |
| 10d | 5.0 | 100 | DMF | 12 | n.d.c |
| 11e | 2.0 | 100 | DMF | 8 | 80 |
| 12f | 2.5 | 80 | DMF | 8 | 63 |
ap-Anisaldehyde (1 mmol) and ARF (0.5 g mmol−1) in DMF or DMA under nitrogen. bYield of isolated product. cNot detected by HPLC. dp-Methoxyacetophenone (1 mmol) and ARF (0.5 g mmol−1). eDichloro(p-cymene)-ruthenium(II) dimer used instead of RuCl3·3H2O. fHCOOK (2 equiv) was used as the reducing source instead of ARF.
The ARF was prepared from commercially available Amberlite® resin (chloride form) by exchanging the anion (chloride) with formic acid following our procedure [24]. A wide range of aryl aldehydes were subjected to reduction under the optimized conditions. Aryl aldehydes substituted with various electron withdrawing and donating groups did not seem to influence the reduction rate as revealed by the similarity of the results and all gave the corresponding alcohols in high yields (Table 2). Several potentially reducible groups such as halogens, nitro etc. were not affected under the reaction conditions (Table 2, entries 6–11, 17). Aliphatic aldehydes (Table 2, entries 14, 15) were also reduced to corresponding alcohols efficiently. Furthermore, the presence of ortho-substituents did not hinder the rate of the reduction as manifested from the reaction conditions (Table 2, entries 6, 9). Hetero-aryl aldehydes were also reduced to corresponding alcohols efficiently (Table 2, entries 16, 17). Surprisingly, aryl ketones were not reduced under similar conditions despite a great deal of variation in experimental conditions (addition of bases, phosphine ligands and application of higher temperatures up to 120 °C). The selectivity between aryl aldehyde and aryl ketone might offer a distinct advantage when both the functional groups are present. Accordingly, we applied the protocol to a mixture of an aryl aldehyde and aryl ketone (1 mmol each). After conducting the reaction at 85 °C for 8 h, the aryl ketone was recovered almost quantitatively along with the reduced product of the aldehyde (Table 2, entry 18). Distinct advantages of cleaner reaction and easy isolation of the product are notable features when comparing the application of heterogeneous ARF and a simple formate salt (herein potassium formate) in homogeneous phase (Table 1, entry 12). The resin beads obtained after filtration from the reaction mixture could be reused for further hydrogenation reactions after washing with methanol and recharging with formic acid.
Table 2: Reduction of aryl aldehydes using resin-supported formate and catalytic RuCl3·3H2O.
| Entry | Substrate | Product |
Conditionsa
Temp / Time |
Yield (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-4-53-i3.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-4-53-i4.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 91 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-4-53-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-4-53-i6.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 74 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-4-53-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-4-53-i8.svg?max-width=637&scale=1.0)
|
85 °C / 9 h | 83 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-4-53-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-4-53-i10.svg?max-width=637&scale=1.0)
|
85 °C / 8 h | 83 |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-4-53-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-4-53-i12.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 70 |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-4-53-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-4-53-i14.svg?max-width=637&scale=1.0)
|
90 °C / 7 h | 78 |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-4-53-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-4-53-i16.svg?max-width=637&scale=1.0)
|
85 °C / 8 h | 70 |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-4-53-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-4-53-i18.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 96 |
| 9 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-4-53-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-4-53-i20.svg?max-width=637&scale=1.0)
|
90 °C / 9 h | 84 |
| 10 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-4-53-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-4-53-i22.svg?max-width=637&scale=1.0)
|
85 °C / 8 h | 72 |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-4-53-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-4-53-i24.svg?max-width=637&scale=1.0)
|
90 °C / 8 h | 79 |
| 12 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-4-53-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-4-53-i26.svg?max-width=637&scale=1.0)
|
90 °C / 8 h | 81 |
| 13 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-4-53-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-4-53-i28.svg?max-width=637&scale=1.0)
|
90 °C / 8 h | 86 |
| 14 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-4-53-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-4-53-i30.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 79 |
| 15 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-4-53-i31.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-4-53-i32.svg?max-width=637&scale=1.0)
|
80 °C / 12 h | 94 |
| 16 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-4-53-i33.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-4-53-i34.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 83 |
| 17 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-4-53-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 34]](/bjoc/content/inline/1860-5397-4-53-i36.svg?max-width=637&scale=1.0)
|
80 °C / 8 h | 76 |
| 18 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-4-53-i37.svg?max-width=637&scale=1.0)
|
![[Graphic 36]](/bjoc/content/inline/1860-5397-4-53-i38.svg?max-width=637&scale=1.0)
|
85 °C / 8 h | 89c |
aAldehyde/ARF/RuCl3·3H2O (1 mmol:500 mg:0.025 mmol) in 2 ml DMF (or DMA). bIsolated yields are average of two runs and alcohols are characterised by spectral data. cNearly quantitative recovery of ketone.
The reaction conditions appear to be mild and base-free, and give high yields of the corresponding alcohols and free of any by-product. Of interest is that, although the use of base co-catalysts for metal complex catalyzed hydrogen transfer is common [27-30], the present reaction conditions without any base preclude possibilities of unwanted reactions of aryl aldehydes, e.g. Cannizzaro reaction.
To broaden the scope of the catalytic system, we tested CTH of 1,2-diketones under similar conditions (Scheme 2). Whereas aryl ketones were not reduced under the conditions, reduction of benzil to benzoin proceeded smoothly in good to excellent yields. Until now, various procedures [31,32] including Lewis acid-mediated conditions [32] have been developed for the reduction of 1,2-dicarbonyl compounds to yield the α-hydroxy ketones without over reduction to diols. The direct use of catalytic RuCl3·3H2O in combination with ARF under neutral conditions could be of interest. Diketones with other substituents also worked efficiently and the results are presented in Scheme 2.
![[1860-5397-4-53-i2]](/bjoc/content/inline/1860-5397-4-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, this system i.e. ARF–RuCl3–DMF (or DMA), constitutes an efficient and selective reductant for reduction of aryl aldehydes and 1,2-diketones to aryl alcohols and α-hydroxy ketones respectively under mild, base-free and phosphine- or any ligand-free conditions. It is observed that aryl ketones and several other potentially reducible functionalities remain unchanged under the reaction conditions. The catalytic system should find further applications since no specially designed chelating ligand-based Ru-complexes are required and the resin-supported H-source (ARF) is easy to prepare and can be stored at room temperature for several months without special precautions.
Experimental
A representative procedure for RuCl3·3H2O-catalyzed transfer hydrogenation of aryl aldehyde using ARF: 1-Naphthaldehyde (156 mg, 1 mmol), ARF (500 mg), RuCl3·3H2O (6.5 mg, 2.5 mol%) and DMF (2 mL) were placed in a screw-capped tube and heated in an oil bath at 80 °C for 8 h. The mixture was cooled, diluted with water (4 mL) and then the resins were filtered off by passing through a cotton bed. The filtrate was diluted with water, extracted with ether (2 × 10 mL) and the combined organic layers were washed with brine and dried over Na2SO4. Removal of the solvent afforded an oil, which was purified through a small pad of silica gel (mesh size 60–120) using ethyl acetate/light petroleum (1:4) to give 1-naphthylmethanol as a colorless solid (144 mg, 91% yield); mp 59–60 °C (Lit. [33] mp 60–62 °C), FT-IR (Nujol): νmax 3317, 2877, 1596, 1512 cm−1; 1H NMR (CDCl3, 300 MHz): δ 8.07–8.04 (m, 1H), 7.86–7.76 (m, 2H), 7.51–7.31 (m, 4H), 5.07 (s, 2H), 2.14 (br s, 1H); 13C NMR (CDCl3, 75 MHz): δ 136.3, 133.8, 131.2, 128.7, 128.6, 126.3, 125.9, 125.4, 125.3, 123.7, 63.6.
Representative procedure for RuCl3·3H2O-catalyzed transfer hydrogenation of aryl aldehyde using HCOOK: 1-Naphthaldehyde (156 mg, 1 mmol), HCOOK (168 mg, 2 mmol), RuCl3·3H2O (6.5 mg, 2.5 mol%) and DMF (2 mL) were placed in a screw-capped tube and heated in an oil bath at 80 °C for 8 h. The mixture was cooled and diluted with water (4 mL) followed by extraction with ether (2 × 10 mL). The combined organic extracts were then washed with brine and dried over anhydrous Na2SO4. Removal of the solvent afforded an oil, which after purification by column chromatography through a small pad of silica gel (mesh size 60–120) using ethyl acetate/light petroleum (1:4) gave 1-naphthylmethanol as a colorless solid (100 mg, 63% yield); mp 57–59 °C.
Supporting Information
Supporting information features general experimental procedures and IR, 1H and 13C NMR spectral data for alcohols (Table 2, entries 3, 5, 6, 8–14, 16, 17) and HRMS data for alcohols (Table 2, entries 3, 12, 13, 14).
| Supporting Information File 1: General experimental procedure | ||
| Format: PDF | Size: 17.8 KB | Download |
| Supporting Information File 2: Spectral data of some selected alcohols | ||
| Format: PDF | Size: 111.7 KB | Download |
References
-
Gladiali, S.; Alberico, E. Chem. Soc. Rev. 2006, 35, 226–236. doi:10.1039/b513396c
Return to citation in text: [1] -
Samec, J. S. M.; Bäckvall, J.-E.; Andersson, P. G.; Brandt, P. Chem. Soc. Rev. 2006, 35, 237–248. doi:10.1039/b515269k
Return to citation in text: [1] -
de Graauw, C. F.; Peters, J. A.; van Bekkum, H.; Huskens, J. Synthesis 1994, 1007–1017. doi:10.1055/s-1994-25625
Return to citation in text: [1] -
Braude, E. A.; Linstead, R. P. J. Chem. Soc. 1954, 3544–3547. doi:10.1039/jr9540003544
Return to citation in text: [1] -
Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97–102. doi:10.1021/ar9502341
And references therein.
Return to citation in text: [1] [2] -
Noyori, R.; Yamakawa, M.; Hashiguchi, S. J. Org. Chem. 2001, 66, 7931–7944. doi:10.1021/jo010721w
Return to citation in text: [1] -
Berthold, H.; Schotten, T.; Hönig, H. Synthesis 2002, 1607–1610. doi:10.1055/s-2002-33349
Return to citation in text: [1] -
Noyori, R.; Takaya, H. Acc. Chem. Res. 1990, 23, 345–350. doi:10.1021/ar00178a005
Return to citation in text: [1] -
Ram, S.; Ehrenkaufer, R. E. Synthesis 1988, 91–95. doi:10.1055/s-1988-27478
Return to citation in text: [1] -
Khai, B. T.; Arcelli, A. Tetrahedron Lett. 1985, 26, 3365–3368. doi:10.1016/S0040-4039(00)98299-6
Return to citation in text: [1] -
Watanabe, Y.; Ohta, T.; Tsuji, Y. Bull. Chem. Soc. Jpn. 1982, 55, 2441–2444. doi:10.1246/bcsj.55.2441
Return to citation in text: [1] -
Halpern, J.; Harrod, J. F.; James, B. R. J. Am. Chem. Soc. 1966, 88, 5150–5155. doi:10.1021/ja00974a022
Return to citation in text: [1] -
James, B. R. Homogeneous Hydrogenation; John Wiley & Sons: New York, 1973; p 94.
Return to citation in text: [1] -
Rebouças, J. S.; James, B. R. Tetrahedron Lett. 2006, 47, 5119–5122. doi:10.1016/j.tetlet.2006.05.083
Return to citation in text: [1] -
Dalal, M. K.; Ram, R. N. Bull. Mater. Sci. 2001, 24, 237–241. doi:10.1007/BF02710108
Return to citation in text: [1] -
Venkatachalam, G.; Ramesh, R. Tetrahedron Lett. 2005, 46, 5215–5218. doi:10.1016/j.tetlet.2005.05.116
Return to citation in text: [1] -
Venkatachalam, G.; Ramesh, R. Inorg. Chem. Commun. 2005, 8, 1009–1013. doi:10.1016/j.inoche.2005.08.004
Return to citation in text: [1] -
Venkatachalam, G.; Ramesh, R.; Mobin, S. M. J. Organomet. Chem. 2005, 690, 3937–3945. doi:10.1016/j.jorganchem.2005.05.039
Return to citation in text: [1] -
Kannan, S.; Ramesh, R.; Liu, Y. J. Organomet. Chem. 2007, 692, 3380–3391. doi:10.1016/j.jorganchem.2007.04.042
Return to citation in text: [1] -
Govindaswamy, P.; Canivet, J.; Therrien, B.; Süss-Fink, G.; Štěpnička, P.; Ludvík, J. J. Organomet. Chem. 2007, 692, 3664–3675. doi:10.1016/j.jorganchem.2007.04.048
Return to citation in text: [1] -
Ley, S. V.; Baxendale, I. R.; Bream, R. N.; Jackson, P. S.; Leach, A. G.; Longbottom, D. A.; Nesi, M.; Scott, J. S.; Storer, R. I.; Taylor, S. J. J. Chem. Soc., Perkin Trans. 1 2000, 3815–4195. doi:10.1039/b006588i
Return to citation in text: [1] -
Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem., Int. Ed. 2001, 40, 650–679. doi:10.1002/1521-3773(20010216)40:4<650::AID-ANIE6500>3.0.CO;2-C
Return to citation in text: [1] -
Basu, B.; Das, P.; Das, S. Mol. Diversity 2005, 9, 259–262. doi:10.1007/s11030-005-8106-1
Return to citation in text: [1] -
Basu, B.; Bhuiyan, M. M. H.; Das, P.; Hossain, I. Tetrahedron Lett. 2003, 44, 8931–8934. doi:10.1016/j.tetlet.2003.10.019
Return to citation in text: [1] [2] -
Desai, B.; Danks, T. N. Tetrahedron Lett. 2001, 42, 5963–5965. doi:10.1016/S0040-4039(01)01157-1
Return to citation in text: [1] -
Abiraj, K.; Srinivasa, G. R.; Gowda, D. C. Synth. Commun. 2005, 35, 223–230. doi:10.1081/SCC-200048429
Return to citation in text: [1] -
Matsumura, K.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1997, 119, 8738–8739. doi:10.1021/ja971570a
Return to citation in text: [1] -
Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 285–288. doi:10.1002/anie.199702851
Return to citation in text: [1] -
Chowdhury, R. L.; Bäckvall, J.-E. J. Chem. Soc., Chem. Commun. 1991, 1063–1064. doi:10.1039/C39910001063
Return to citation in text: [1] -
Mezzetti, A.; Consiglio, G. J. Chem. Soc., Chem. Commun. 1991, 1675–1677. doi:10.1039/C39910001675
Return to citation in text: [1] -
Hayakawa, R.; Sahara, T.; Shimizu, M. Tetrahedron Lett. 2000, 41, 7939–7942. doi:10.1016/S0040-4039(00)01385-X
Return to citation in text: [1] -
Kikuchi, S.; Hashimoto, Y. Synlett 2004, 1267–1269. doi:10.1055/s-2004-825581
Return to citation in text: [1] [2] -
Buckingham, J., Ed. Dictionary of Organic Compounds, 5th ed.; Chapman & Hall: New York, 1982; Vol. 4, p 4186.
Return to citation in text: [1]
| 32. | Kikuchi, S.; Hashimoto, Y. Synlett 2004, 1267–1269. doi:10.1055/s-2004-825581 |
| 33. | Buckingham, J., Ed. Dictionary of Organic Compounds, 5th ed.; Chapman & Hall: New York, 1982; Vol. 4, p 4186. |
| 1. | Gladiali, S.; Alberico, E. Chem. Soc. Rev. 2006, 35, 226–236. doi:10.1039/b513396c |
| 2. | Samec, J. S. M.; Bäckvall, J.-E.; Andersson, P. G.; Brandt, P. Chem. Soc. Rev. 2006, 35, 237–248. doi:10.1039/b515269k |
| 3. | de Graauw, C. F.; Peters, J. A.; van Bekkum, H.; Huskens, J. Synthesis 1994, 1007–1017. doi:10.1055/s-1994-25625 |
| 5. |
Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97–102. doi:10.1021/ar9502341
And references therein. |
| 27. | Matsumura, K.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1997, 119, 8738–8739. doi:10.1021/ja971570a |
| 28. | Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 285–288. doi:10.1002/anie.199702851 |
| 29. | Chowdhury, R. L.; Bäckvall, J.-E. J. Chem. Soc., Chem. Commun. 1991, 1063–1064. doi:10.1039/C39910001063 |
| 30. | Mezzetti, A.; Consiglio, G. J. Chem. Soc., Chem. Commun. 1991, 1675–1677. doi:10.1039/C39910001675 |
| 7. | Berthold, H.; Schotten, T.; Hönig, H. Synthesis 2002, 1607–1610. doi:10.1055/s-2002-33349 |
| 8. | Noyori, R.; Takaya, H. Acc. Chem. Res. 1990, 23, 345–350. doi:10.1021/ar00178a005 |
| 9. | Ram, S.; Ehrenkaufer, R. E. Synthesis 1988, 91–95. doi:10.1055/s-1988-27478 |
| 10. | Khai, B. T.; Arcelli, A. Tetrahedron Lett. 1985, 26, 3365–3368. doi:10.1016/S0040-4039(00)98299-6 |
| 11. | Watanabe, Y.; Ohta, T.; Tsuji, Y. Bull. Chem. Soc. Jpn. 1982, 55, 2441–2444. doi:10.1246/bcsj.55.2441 |
| 31. | Hayakawa, R.; Sahara, T.; Shimizu, M. Tetrahedron Lett. 2000, 41, 7939–7942. doi:10.1016/S0040-4039(00)01385-X |
| 32. | Kikuchi, S.; Hashimoto, Y. Synlett 2004, 1267–1269. doi:10.1055/s-2004-825581 |
| 5. |
Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97–102. doi:10.1021/ar9502341
And references therein. |
| 6. | Noyori, R.; Yamakawa, M.; Hashiguchi, S. J. Org. Chem. 2001, 66, 7931–7944. doi:10.1021/jo010721w |
| 26. | Abiraj, K.; Srinivasa, G. R.; Gowda, D. C. Synth. Commun. 2005, 35, 223–230. doi:10.1081/SCC-200048429 |
| 4. | Braude, E. A.; Linstead, R. P. J. Chem. Soc. 1954, 3544–3547. doi:10.1039/jr9540003544 |
| 24. | Basu, B.; Bhuiyan, M. M. H.; Das, P.; Hossain, I. Tetrahedron Lett. 2003, 44, 8931–8934. doi:10.1016/j.tetlet.2003.10.019 |
| 16. | Venkatachalam, G.; Ramesh, R. Tetrahedron Lett. 2005, 46, 5215–5218. doi:10.1016/j.tetlet.2005.05.116 |
| 17. | Venkatachalam, G.; Ramesh, R. Inorg. Chem. Commun. 2005, 8, 1009–1013. doi:10.1016/j.inoche.2005.08.004 |
| 18. | Venkatachalam, G.; Ramesh, R.; Mobin, S. M. J. Organomet. Chem. 2005, 690, 3937–3945. doi:10.1016/j.jorganchem.2005.05.039 |
| 19. | Kannan, S.; Ramesh, R.; Liu, Y. J. Organomet. Chem. 2007, 692, 3380–3391. doi:10.1016/j.jorganchem.2007.04.042 |
| 20. | Govindaswamy, P.; Canivet, J.; Therrien, B.; Süss-Fink, G.; Štěpnička, P.; Ludvík, J. J. Organomet. Chem. 2007, 692, 3664–3675. doi:10.1016/j.jorganchem.2007.04.048 |
| 23. | Basu, B.; Das, P.; Das, S. Mol. Diversity 2005, 9, 259–262. doi:10.1007/s11030-005-8106-1 |
| 24. | Basu, B.; Bhuiyan, M. M. H.; Das, P.; Hossain, I. Tetrahedron Lett. 2003, 44, 8931–8934. doi:10.1016/j.tetlet.2003.10.019 |
| 15. | Dalal, M. K.; Ram, R. N. Bull. Mater. Sci. 2001, 24, 237–241. doi:10.1007/BF02710108 |
| 25. | Desai, B.; Danks, T. N. Tetrahedron Lett. 2001, 42, 5963–5965. doi:10.1016/S0040-4039(01)01157-1 |
| 14. | Rebouças, J. S.; James, B. R. Tetrahedron Lett. 2006, 47, 5119–5122. doi:10.1016/j.tetlet.2006.05.083 |
| 12. | Halpern, J.; Harrod, J. F.; James, B. R. J. Am. Chem. Soc. 1966, 88, 5150–5155. doi:10.1021/ja00974a022 |
| 13. | James, B. R. Homogeneous Hydrogenation; John Wiley & Sons: New York, 1973; p 94. |
| 21. | Ley, S. V.; Baxendale, I. R.; Bream, R. N.; Jackson, P. S.; Leach, A. G.; Longbottom, D. A.; Nesi, M.; Scott, J. S.; Storer, R. I.; Taylor, S. J. J. Chem. Soc., Perkin Trans. 1 2000, 3815–4195. doi:10.1039/b006588i |
| 22. | Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem., Int. Ed. 2001, 40, 650–679. doi:10.1002/1521-3773(20010216)40:4<650::AID-ANIE6500>3.0.CO;2-C |
© 2008 Basu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)