Abstract
A miniflow system for oxidative cyclization of alkenols with Oxone was developed. Thus, the oxidative cyclization of (Z)- and (E)-alkenols in i-PrOH with an aqueous solution of Oxone proceeded smoothly and safely in a PTFE tube without any exogenous catalytic species, and was subsequently quenched in a flow-reaction manner to afford the corresponding furanyl and pyranyl carbinols quantitatively within 5 or 10 min of residence time.

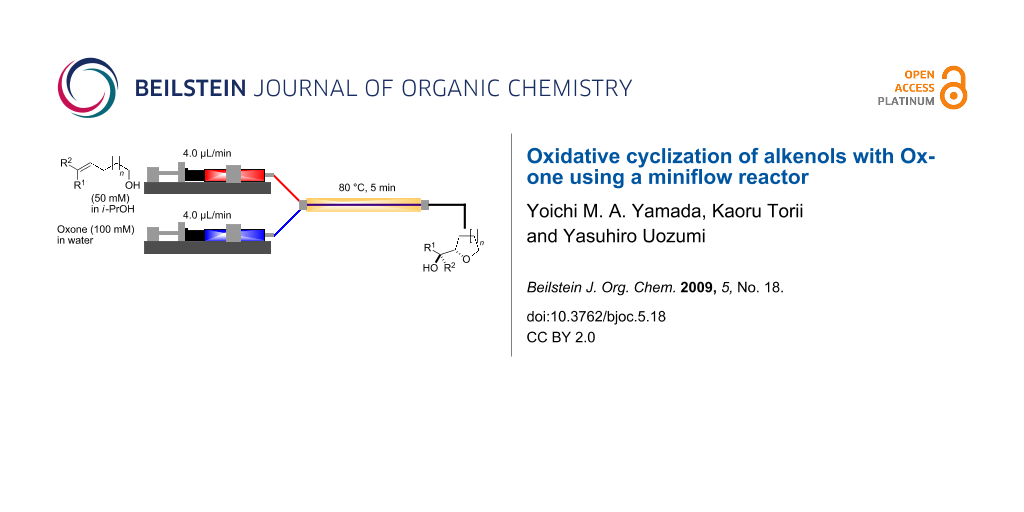
Graphical Abstract
Introduction
The development of flow-reaction systems for molecular transformations is an important goal in organic syntheses. Recently, innovative devices such as micro- and miniflow reactors that offer many fundamental as well as practical advantages for efficient organic transformations have been gaining ground in chemical experimentation [1-15]. Extensive investigations have revealed that the large interfacial area and the short molecular diffusion path in narrow space reactors often drastically improve the efficiency of a given chemical reaction. As a case in point, we have previously developed a catalyst-installed microflow reactor where a membranous polymeric palladium catalyst was deposited inside a micro-channel reactor at the laminar flow interface [16], resulting in the instantaneous production of biaryls (quantitative yield within 4 s of residence time) via a palladium-catalyzed Suzuki-Miyaura reaction under microflow conditions. An additional advantage of micro- and minireactors is the small heat capacity of the micro- and miniflow systems thus rendering exothermic and/or potentially explosive reactions safe and practical. Consequently, oxidative transformations with potentially explosive oxidants would be ideal target reactions for miniflow systems. We wish to report the oxidative construction of furanyl and pyranyl alkyl carbinols with Oxone via a miniflow reaction system.
Results and Discussion
Furanyl and pyranyl carbinols have generated considerable interest due to their presence in a number of therapeutically and biologically active compounds [17-31]. We therefore decided to turn our attention to developing an oxidative cyclization of alkenols [32,33] for the preparation of furanyl and pyranyl alkyl carbinols. During our investigation, we found that the oxidative cyclization of (Z)-4-decen-1-ol (1a) with Oxone (2KHSO5·KHSO4·K2SO4) proceeded at 80 °C without any exogenous catalysts under small-scale batch conditions (up to 50 mmol of 1a) to give threo-1-(2-tetrahydrofuranyl)hexan-1-ol (2a) in 99% yield within 5 min (Table 1) [34]. When a mixture of an aqueous solution of Oxone (100 mM, 1 mL, 2 equiv vs 1a) and a 2-propanol solution of 1a (50 mM, 1 mL) was stirred at 80 °C for 5 min, the cyclization took place very smoothly to afford threo-1-(2-tetrahydrofuranyl)hexan-1-ol (2a) in 99% yield as a single racemic diastereoisomer. Yet when 30% aq H2O2 was used as the oxidant at 80 °C, the cyclization hardly proceeded at all, even with a longer reaction time [32]. We had previously found that a polymeric phosphotungstate catalyst promoted the cyclization of 1a with 30% aq H2O2 at 50 °C with a much longer reaction time (24 h). Thus, Oxone was found to be the most efficient oxidant to promote the cyclization of (Z)-4-decen-1-ol (1a). Although a powerful and inexpensive oxidant for this transformation [35-42], Oxone however is also a known fire and explosion hazard [43] essentially rendering its large-scale use impractical. To avoid these potentially dangerous and hazardous conditions in a large-scale batch oxidation, we switched the conventional batch system to a miniflow system.
Table 1: The oxidative cyclization of an alkenol 1a with Oxone under batch conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-5-18-i1.svg?max-width=637&scale=1.0)
|
||
| oxidant | conditions | yield of 2a (%) |
|---|---|---|
| aq Oxone | 80 °C, 5 min | 99 |
| 30% aq H2O2 | 80 °C, 60 min | no reaction |
| polymeric PW12O403− (cat) with 30% aq H2O2 (see [32]) | 50 °C, 24 h | 99 |
The miniflow reaction system is composed of poly(tetrafluoroethylene) (PTFE) tubes of ø = 1 mm, T-shaped connectors, and syringes with syringe pumps as shown in Figure 1. When the miniflow reaction of the alkenols 1 in i-PrOH with an aqueous solution of Oxone was carried out in the miniflow reactor with 5 min of residence time at 80 °C, we were pleased to see that the reaction proceeded smoothly to afford the corresponding cyclic ethers 2 in high conversion. Thus, a solution of an alkenol 1 in i-PrOH (50 mM) and Oxone in water (100 mM) were oppositely injected with a flow rate of 4.0 μl/min each by using syringe pumps from the individual inlets. The mixed solution passed through a PTFE tube reactor (length = 50 mm) at 80 °C, and then was quenched with 30% aq Na2S2O3 solution injected into the flow tube with a flow rate of 4.0 μl/min. The resulting organic/aqueous outflow was collected in a glass vial. The chemical conversion and structure of the products were determined by GC and 1H NMR analysis. As shown in Table 2, entry 1, the oxidative cyclization of (Z)-4-decen-1-ol (1a) with Oxone was performed within 5 min of residence time to afford threo-1-(2-tetrahydrofuranyl)hexan-1-ol (2a) in 99% conversion. The cyclization of (Z)-4-hexen-1-ol (1b) and (Z)-4-hepten-1-ol (1c) proceeded smoothly to give the threo-tetrahydrofuranyl alcohols 2b and 2c in 90% and 88% conversion, respectively (entries 2 and 3). This flow reaction system was also utilized for the formation of six-membered cyclic ethers. Thus, the oxidation of (Z)-5-octen-1-ol (1d) was carried out with a flow rate of 2.0 μL/min (residence time: 10 min) to give 90% conversion of the threo-tetrahydropyranyl alcohol 2d (entry 4). (E)-4-Decen-1-ol (1e) underwent oxidative cyclization with 10 min residence time to afford the erythro-product 2e in 70% conversion (entry 5). These stereochemical observations indicate that the cyclization involves a stereospecific reaction pathway. The reaction pathway of the present oxidative cyclization should proceed via the epoxidation of the alkene 1 with Oxone and subsequent oxirane ring opening with the intramolecular oxygen nucleophile (an intramolecular SN2 reaction) to afford the product 2 stereospecifically [44,45]. It should be noted that the miniflow cyclization of 1a was continuously carried out to give a quantitative conversion of 2a over 2 h.
![[1860-5397-5-18-1]](/bjoc/content/figures/1860-5397-5-18-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Miniflow reaction system of oxidative cyclization.
Figure 1: Miniflow reaction system of oxidative cyclization.
Table 2: Oxidative cyclization of alkenols with Oxone through a miniflow reactor.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-18-i2.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Conversion (%) |
|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-5-18-i3.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-5-18-i4.svg?max-width=637&scale=1.0)
2a |
99 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-5-18-i5.svg?max-width=637&scale=1.0)
1b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-5-18-i6.svg?max-width=637&scale=1.0)
2b |
90 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-5-18-i7.svg?max-width=637&scale=1.0)
1c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-5-18-i8.svg?max-width=637&scale=1.0)
2c |
88 |
| 4b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-5-18-i9.svg?max-width=637&scale=1.0)
1d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-5-18-i10.svg?max-width=637&scale=1.0)
2d |
90 |
| 5b |
![[Graphic 11]](/bjoc/content/inline/1860-5397-5-18-i11.svg?max-width=637&scale=1.0)
1e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-5-18-i12.svg?max-width=637&scale=1.0)
2e |
70 |
aalkenol (50 mM in i-PrOH), Oxone (2KHSO5·KHSO4·K2SO4) (100 mM in H2O), flow rate: 4.0 μL/min each, 80 °C, residence time = 5 min; several fractions were collected for each reaction shown to demonstrate the stable and high reactive performance of the miniflow reactor; the product 2a was obtained in 0.12 mmol/h. bflow rate: 2.0 μL/min each, 80 °C, residence time = 10 min.
Conclusion
In conclusion, we have developed a miniflow reaction system for the oxidative cyclization of alkenols with Oxone, affording the corresponding cyclic ethers in high conversion, where potentially explosive Oxone was used and quenched safely. Development of instantaneous flow reaction systems for the oxidation reactions with a retention time of several seconds is currently in progress.
Acknowledgements
This work was supported by the GSC project, sponsored by the METI. We thank the JSPS (Grant-in-Aid for Scientific Research, no.15205015, no. 16790025, no.18065019 and no. 20655035), and the MEXT (Scientific Research on Priority Areas, no. 460) for partial financial support of this work.
References
-
Yoshida, J.-i.; Nagaki, A.; Yamada, T. Chem.–Eur. J. 2008, 14, 7450–7459. doi:10.1002/chem.200800582
(See for reviews.)
Return to citation in text: [1] -
Fukuyama, T.; Rahman, M. T.; Sato, M.; Ryu, I. Synlett 2008, 151–163. doi:10.1055/s-2007-1000884
Return to citation in text: [1] -
Wiles, C.; Watts, P. Eur. J. Org. Chem. 2008, 1655–1671. doi:10.1002/ejoc.200701041
Return to citation in text: [1] -
Kobayashi, J.; Mori, Y.; Kobayashi, S. Chem.–Asian J. 2006, 1, 22–35. doi:10.1002/asia.200600058
Return to citation in text: [1] -
Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577
Return to citation in text: [1] -
Pennemann, H.; Hessel, V.; Löwe, H. Chem. Eng. Sci. 2004, 59, 4789–4794. doi:10.1016/j.ces.2004.07.049
Return to citation in text: [1] -
Fletcher, P. D. I.; Haswell, S. J.; Pombo-Villar, E.; Warrington, B. H.; Watts, P.; Wong, S. Y. F.; Zhang, X. Tetrahedron 2002, 58, 4735–4754. doi:10.1016/S0040-4020(02)00432-5
Return to citation in text: [1] -
Haswell, S. J.; Middleton, R. J.; O’Sullivan, B.; Skelton, V.; Watts, P.; Styring, P. Chem. Commun. 2001, 391–398. doi:10.1039/b008496o
Return to citation in text: [1] -
Fukuyama, T.; Kobayashi, M.; Rahman, M. T.; Kamata, N.; Ryu, I. Org. Lett. 2008, 10, 533–536. doi:10.1021/ol702718z
(See for selected examples.)
Return to citation in text: [1] -
Hornung, C. H.; Mackley, M. R.; Baxendale, I. R.; Ley, S. V. Org. Process Res. Dev. 2007, 11, 399–405. doi:10.1021/op700015f
Return to citation in text: [1] -
Sahoo, H. R.; Kralj, J. G.; Jensen, K. F. Angew. Chem., Int. Ed. 2007, 46, 5704–5708. doi:10.1002/anie.200701434
Return to citation in text: [1] -
Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka, S.; Fukase, K. Org. Lett. 2007, 9, 299–302. doi:10.1021/ol062777o
Return to citation in text: [1] -
He, P.; Watts, P.; Marken, F.; Haswell, S. J. Angew. Chem., Int. Ed. 2006, 45, 4146–4149. doi:10.1002/anie.200600951
Return to citation in text: [1] -
Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 11666–11675. doi:10.1021/ja0527424
Return to citation in text: [1] -
Kawaguchi, T.; Miyata, H.; Ataka, K.; Mae, K.; Yoshida, J. Angew. Chem., Int. Ed. 2005, 44, 2413–2416. doi:10.1002/anie.200462466
(See for Swern oxidations by using a microflow system.)
Return to citation in text: [1] -
Uozumi, Y.; Yamada, Y. M. A.; Beppu, T.; Fukuyama, N.; Ueno, M.; Kitamori, T. J. Am. Chem. Soc. 2006, 128, 15994–15995. doi:10.1021/ja066697r
Return to citation in text: [1] -
Alali, F. Q.; Liu, X.-X.; Mclaughlin, J. L. J. Nat. Prod. 1999, 62, 504–540. doi:10.1021/np980406d
Return to citation in text: [1] -
Hartung, J.; Greb, M. J. Organomet. Chem. 2002, 661, 67–84. doi:10.1016/S0022-328X(02)01807-7
Return to citation in text: [1] -
Nakata, T.; Schmid, G.; Vranesic, B.; Okigawa, M.; Smith-Palmer, T.; Kishi, Y. J. Am. Chem. Soc. 1978, 100, 2933–2935. doi:10.1021/ja00477a081
Return to citation in text: [1] -
Fukuyama, T.; Wang, C.-L. J.; Kishi, Y. J. Am. Chem. Soc. 1979, 101, 260–262. doi:10.1021/ja00495a065
Return to citation in text: [1] -
Wuts, P. G. M.; D’Costa, R.; Butler, W. J. Org. Chem. 1984, 49, 2582–2588. doi:10.1021/jo00188a014
Return to citation in text: [1] -
Still, W. C.; Romero, A. G. J. Am. Chem. Soc. 1986, 108, 2105–2106. doi:10.1021/ja00268a069
Return to citation in text: [1] -
Boivin, T. L. B. Tetrahedron 1987, 43, 3309–3362. doi:10.1016/S0040-4020(01)81626-4
Return to citation in text: [1] -
Evans, D. A.; Polniaszek, R. P.; Devries, K. M.; Guinn, D. E.; Mathre, D. J. J. Am. Chem. Soc. 1991, 113, 7613–7630. doi:10.1021/ja00020a025
Return to citation in text: [1] -
Sinha, S.; Sinha-Bagchi, A.; Keinan, E. J. Am. Chem. Soc. 1995, 117, 1447–1448. doi:10.1021/ja00109a037
Return to citation in text: [1] -
Koert, U. Synthesis 1995, 115–132. doi:10.1055/s-1995-3883
Return to citation in text: [1] -
Sakaguchi, S.; Nishiyama, Y.; Ishii, Y. J. Org. Chem. 1996, 61, 5307–5311. doi:10.1021/jo960275q
Return to citation in text: [1] -
Towne, T. B.; McDonald, F. E. J. Am. Chem. Soc. 1997, 119, 6022–6028. doi:10.1021/ja962837t
Return to citation in text: [1] -
Wang, Z.-M.; Tian, S.-K.; Shi, M. Tetrahedron Lett. 1999, 40, 977–980. doi:10.1016/S0040-4039(98)02577-5
Return to citation in text: [1] -
Bhaumik, A.; Tatsumi, T. J. Catal. 2000, 189, 31–39. doi:10.1006/jcat.1999.2690
Return to citation in text: [1] -
Ichihara, J.; Kambara, A.; Iteya, K.; Sugimoto, E.; Shinkawa, T.; Takaoka, A.; Yamaguchi, S.; Sasaki, Y. Green Chem. 2003, 5, 491–493. doi:10.1039/b303315e
Return to citation in text: [1] -
Yamada, Y. M. A.; Guo, H.; Uozumi, Y. Org. Lett. 2007, 9, 1501–1504. doi:10.1021/ol070258v
Return to citation in text: [1] [2] [3] -
Yamada, Y. M. A.; Guo, H.; Uozumi, Y. Heterocycles 2008, 76, 645–655. doi:10.3987/COM-08-S(N)56
Return to citation in text: [1] -
Yamada, Y. M. A.; Torii, K.; Uozumi, Y. Unpublished results.
Return to citation in text: [1] -
Travis, B. R.; Sivakumar, M.; Hollist, G. O.; Borhan, B. Org. Lett. 2003, 5, 1031–1034. doi:10.1021/ol0340078
(See for examples of batch oxidation reactions with Oxone in the absence of catalysts.)
Return to citation in text: [1] -
Curini, M.; Epifano, F.; Marcotullio, M. C.; Rosati, O. Synlett 1999, 777–779. doi:10.1055/s-1999-2703
Return to citation in text: [1] -
Webb, K. S.; Levy, D. Tetrahedron Lett. 1995, 36, 5117–5118. doi:10.1016/0040-4039(95)00963-D
Return to citation in text: [1] -
Denmark, S. E.; Forbes, D. C.; Hays, D. S.; DePue, J. S.; Wilde, R. G. J. Org. Chem. 1995, 60, 1391–1407. doi:10.1021/jo00110a049
Return to citation in text: [1] -
Webb, S. B. Tetrahedron Lett. 1994, 35, 3457–3460. doi:10.1016/S0040-4039(00)73209-6
Return to citation in text: [1] -
Davis, F. A.; Lal, S. G.; Durst, D. J. Org. Chem. 1988, 53, 5004–5007. doi:10.1021/jo00256a018
Return to citation in text: [1] -
Trost, B. M.; Curran, D. P. Tetrahedron Lett. 1981, 22, 1287–1290. doi:10.1016/S0040-4039(01)90298-9
Return to citation in text: [1] -
Kennedy, R. J.; Stock, A. M. J. Org. Chem. 1960, 25, 1901–1906. doi:10.1021/jo01081a019
Return to citation in text: [1] -
See the MSDS (prepared by Du Pont Chemicals) for Oxone, a registered trademark of Du Pont Chemicals.
Return to citation in text: [1] -
Misono, M. Chem. Commun. 2001, 1141–1152. doi:10.1039/b102573m
Return to citation in text: [1] -
Mizuno, N.; Misono, M. Chem. Lett. 1987, 967–970. doi:10.1246/cl.1987.967
Return to citation in text: [1]
| 1. |
Yoshida, J.-i.; Nagaki, A.; Yamada, T. Chem.–Eur. J. 2008, 14, 7450–7459. doi:10.1002/chem.200800582
(See for reviews.) |
| 2. | Fukuyama, T.; Rahman, M. T.; Sato, M.; Ryu, I. Synlett 2008, 151–163. doi:10.1055/s-2007-1000884 |
| 3. | Wiles, C.; Watts, P. Eur. J. Org. Chem. 2008, 1655–1671. doi:10.1002/ejoc.200701041 |
| 4. | Kobayashi, J.; Mori, Y.; Kobayashi, S. Chem.–Asian J. 2006, 1, 22–35. doi:10.1002/asia.200600058 |
| 5. | Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577 |
| 6. | Pennemann, H.; Hessel, V.; Löwe, H. Chem. Eng. Sci. 2004, 59, 4789–4794. doi:10.1016/j.ces.2004.07.049 |
| 7. | Fletcher, P. D. I.; Haswell, S. J.; Pombo-Villar, E.; Warrington, B. H.; Watts, P.; Wong, S. Y. F.; Zhang, X. Tetrahedron 2002, 58, 4735–4754. doi:10.1016/S0040-4020(02)00432-5 |
| 8. | Haswell, S. J.; Middleton, R. J.; O’Sullivan, B.; Skelton, V.; Watts, P.; Styring, P. Chem. Commun. 2001, 391–398. doi:10.1039/b008496o |
| 9. |
Fukuyama, T.; Kobayashi, M.; Rahman, M. T.; Kamata, N.; Ryu, I. Org. Lett. 2008, 10, 533–536. doi:10.1021/ol702718z
(See for selected examples.) |
| 10. | Hornung, C. H.; Mackley, M. R.; Baxendale, I. R.; Ley, S. V. Org. Process Res. Dev. 2007, 11, 399–405. doi:10.1021/op700015f |
| 11. | Sahoo, H. R.; Kralj, J. G.; Jensen, K. F. Angew. Chem., Int. Ed. 2007, 46, 5704–5708. doi:10.1002/anie.200701434 |
| 12. | Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka, S.; Fukase, K. Org. Lett. 2007, 9, 299–302. doi:10.1021/ol062777o |
| 13. | He, P.; Watts, P.; Marken, F.; Haswell, S. J. Angew. Chem., Int. Ed. 2006, 45, 4146–4149. doi:10.1002/anie.200600951 |
| 14. | Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 11666–11675. doi:10.1021/ja0527424 |
| 15. |
Kawaguchi, T.; Miyata, H.; Ataka, K.; Mae, K.; Yoshida, J. Angew. Chem., Int. Ed. 2005, 44, 2413–2416. doi:10.1002/anie.200462466
(See for Swern oxidations by using a microflow system.) |
| 32. | Yamada, Y. M. A.; Guo, H.; Uozumi, Y. Org. Lett. 2007, 9, 1501–1504. doi:10.1021/ol070258v |
| 33. | Yamada, Y. M. A.; Guo, H.; Uozumi, Y. Heterocycles 2008, 76, 645–655. doi:10.3987/COM-08-S(N)56 |
| 17. | Alali, F. Q.; Liu, X.-X.; Mclaughlin, J. L. J. Nat. Prod. 1999, 62, 504–540. doi:10.1021/np980406d |
| 18. | Hartung, J.; Greb, M. J. Organomet. Chem. 2002, 661, 67–84. doi:10.1016/S0022-328X(02)01807-7 |
| 19. | Nakata, T.; Schmid, G.; Vranesic, B.; Okigawa, M.; Smith-Palmer, T.; Kishi, Y. J. Am. Chem. Soc. 1978, 100, 2933–2935. doi:10.1021/ja00477a081 |
| 20. | Fukuyama, T.; Wang, C.-L. J.; Kishi, Y. J. Am. Chem. Soc. 1979, 101, 260–262. doi:10.1021/ja00495a065 |
| 21. | Wuts, P. G. M.; D’Costa, R.; Butler, W. J. Org. Chem. 1984, 49, 2582–2588. doi:10.1021/jo00188a014 |
| 22. | Still, W. C.; Romero, A. G. J. Am. Chem. Soc. 1986, 108, 2105–2106. doi:10.1021/ja00268a069 |
| 23. | Boivin, T. L. B. Tetrahedron 1987, 43, 3309–3362. doi:10.1016/S0040-4020(01)81626-4 |
| 24. | Evans, D. A.; Polniaszek, R. P.; Devries, K. M.; Guinn, D. E.; Mathre, D. J. J. Am. Chem. Soc. 1991, 113, 7613–7630. doi:10.1021/ja00020a025 |
| 25. | Sinha, S.; Sinha-Bagchi, A.; Keinan, E. J. Am. Chem. Soc. 1995, 117, 1447–1448. doi:10.1021/ja00109a037 |
| 26. | Koert, U. Synthesis 1995, 115–132. doi:10.1055/s-1995-3883 |
| 27. | Sakaguchi, S.; Nishiyama, Y.; Ishii, Y. J. Org. Chem. 1996, 61, 5307–5311. doi:10.1021/jo960275q |
| 28. | Towne, T. B.; McDonald, F. E. J. Am. Chem. Soc. 1997, 119, 6022–6028. doi:10.1021/ja962837t |
| 29. | Wang, Z.-M.; Tian, S.-K.; Shi, M. Tetrahedron Lett. 1999, 40, 977–980. doi:10.1016/S0040-4039(98)02577-5 |
| 30. | Bhaumik, A.; Tatsumi, T. J. Catal. 2000, 189, 31–39. doi:10.1006/jcat.1999.2690 |
| 31. | Ichihara, J.; Kambara, A.; Iteya, K.; Sugimoto, E.; Shinkawa, T.; Takaoka, A.; Yamaguchi, S.; Sasaki, Y. Green Chem. 2003, 5, 491–493. doi:10.1039/b303315e |
| 16. | Uozumi, Y.; Yamada, Y. M. A.; Beppu, T.; Fukuyama, N.; Ueno, M.; Kitamori, T. J. Am. Chem. Soc. 2006, 128, 15994–15995. doi:10.1021/ja066697r |
| 32. | Yamada, Y. M. A.; Guo, H.; Uozumi, Y. Org. Lett. 2007, 9, 1501–1504. doi:10.1021/ol070258v |
| 43. | See the MSDS (prepared by Du Pont Chemicals) for Oxone, a registered trademark of Du Pont Chemicals. |
| 35. |
Travis, B. R.; Sivakumar, M.; Hollist, G. O.; Borhan, B. Org. Lett. 2003, 5, 1031–1034. doi:10.1021/ol0340078
(See for examples of batch oxidation reactions with Oxone in the absence of catalysts.) |
| 36. | Curini, M.; Epifano, F.; Marcotullio, M. C.; Rosati, O. Synlett 1999, 777–779. doi:10.1055/s-1999-2703 |
| 37. | Webb, K. S.; Levy, D. Tetrahedron Lett. 1995, 36, 5117–5118. doi:10.1016/0040-4039(95)00963-D |
| 38. | Denmark, S. E.; Forbes, D. C.; Hays, D. S.; DePue, J. S.; Wilde, R. G. J. Org. Chem. 1995, 60, 1391–1407. doi:10.1021/jo00110a049 |
| 39. | Webb, S. B. Tetrahedron Lett. 1994, 35, 3457–3460. doi:10.1016/S0040-4039(00)73209-6 |
| 40. | Davis, F. A.; Lal, S. G.; Durst, D. J. Org. Chem. 1988, 53, 5004–5007. doi:10.1021/jo00256a018 |
| 41. | Trost, B. M.; Curran, D. P. Tetrahedron Lett. 1981, 22, 1287–1290. doi:10.1016/S0040-4039(01)90298-9 |
| 42. | Kennedy, R. J.; Stock, A. M. J. Org. Chem. 1960, 25, 1901–1906. doi:10.1021/jo01081a019 |
| 32. | Yamada, Y. M. A.; Guo, H.; Uozumi, Y. Org. Lett. 2007, 9, 1501–1504. doi:10.1021/ol070258v |
| 44. | Misono, M. Chem. Commun. 2001, 1141–1152. doi:10.1039/b102573m |
| 45. | Mizuno, N.; Misono, M. Chem. Lett. 1987, 967–970. doi:10.1246/cl.1987.967 |
© 2009 Yamada et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)