Abstract
The first direct synthesis of β-unsubstituted meso-decamethylcalix[5]pyrrole from pyrrole and acetone, with moderate yield, is described. The results showed that a bismuth salt was necessary to obtain calix[5]pyrrole, with the best results obtained using Bi(NO3)3.

Graphical Abstract
Results and Discussion
Calix[n]pyrroles have attracted attention because of their ability to recognize anions [1,2]. To date, the calix[4]pyrroles have been studied the most, in part due to the ease with which the macrocycle can be obtained by the condensation of pyrrole with a ketone catalyzed by a Brønsted-Lowry acid such as HCl or methanesulfonic acid, or a Lewis acid such as zeolites with aluminium or cobalt, BF3 or a bismuth salt [2-5]. The synthesis of calix[n]pyrroles where n > 4 has been reported for n = 5 or 6. The latter compounds have been synthesized via two routes: a) from the sterically hindered diaryldi(pyrrol-2-yl)methane with 25% yield; and b) through the conversion of a calix[6]furan into the corresponding calix[6]pyrrole by an opening process of the six heterocycles, a selective reduction of the double bond and then a Paal-Knorr condensation with ammonium acetate with 40% yield [6,7]. On the other hand, β-unsubstituted calix[5]pyrroles have been obtained by two routes: a) from the corresponding meso-decamethylcalix[5]furan, via a method analogous to that reported for calix[6]pyrroles, with 1% yield; and b) directly when the macrocycle is covalently bound to a calix[5]arene, with 10% yield [8,9]. However, these approaches afford calix[5]pyrroles in low yield, which has limited the study of these compounds as anion receptors.
One explanation for why it is difficult to obtain calix[5]pyrroles via direct condensation of a pyrrole and the corresponding ketone is that the five heterocycle system is unstable: it opens and loses a pyrrole-isopropyl fragment to give the calix[4]pyrrole [8,10].
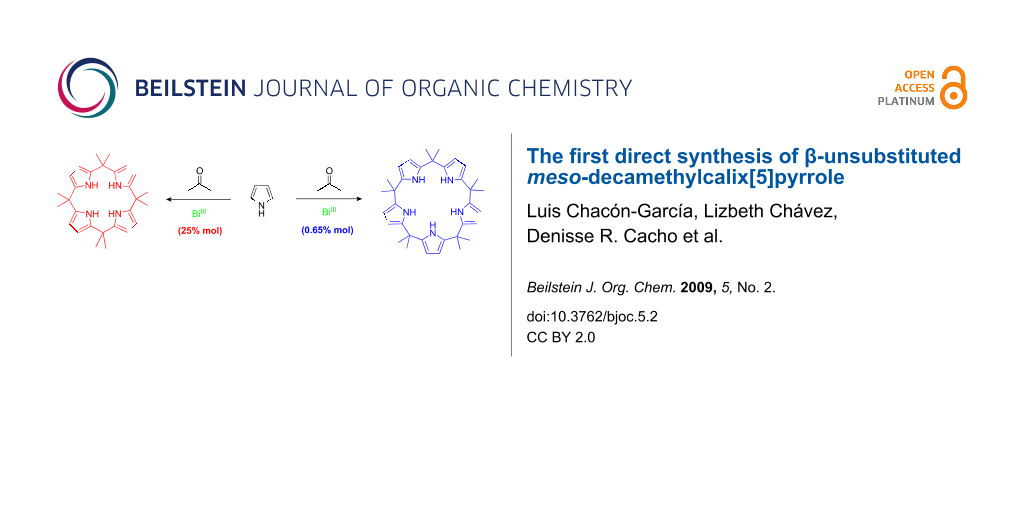
In a recent report we described the synthesis of calix[4]pyrroles via the direct condensation of pyrrole with a series of ketones in the presence of a bismuth salt such as Bi(NO3)3, BiCl3, BiI3, and Bi(CF3SO3)3, in a 1 : 1 : 0.25 (pyrrole : ketone : BiX3) ratio or with the ketone as a solvent at room temperature [5]. Here we describe the first direct synthesis of β-unsubstituted meso-decamethylcalix[5]pyrrole (2) with Bi(NO3)3 in moderate yield (Scheme 1).
![[1860-5397-5-2-i1]](/bjoc/content/inline/1860-5397-5-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Products obtained by the reaction of pyrrole and acetone with bismuth(III).
Scheme 1: Products obtained by the reaction of pyrrole and acetone with bismuth(III).
While studying the role of bismuth as a Lewis acid in the synthesis of calix[4]pyrroles, we found that at low catalyst concentrations some additional products were formed, as observed by 1H NMR spectroscopy. These byproducts exhibited 1H NMR, 13C NMR and MS data consistent with those reported for calix[n]pyrroles with n = 4, 5 and 6 (compounds 1–3, respectively) and 5,5-dimethyldipyrromethane (4); see Experimental section [5,6,8]. The relative proportions of these four products obtained using different catalyst equivalents are listed in Table 1. Compounds 1 and 2 were almost indistinguishable on TLC because of their similar Rf values, and recrystallization from ethanol, as reported in other works, was not satisfactory to give the pure compounds. However, it was possible to separate 1 and 2 by HPLC, to obtain 2 in 25% yield (using the conditions specified in Table 1, entry 12). Compound 2 was found to be unstable, which probably decreased the yield.
Table 1: Catalyst conditions and relative proportions of compounds 1, 2, 3 and 4 detected in the crude reaction mixture by 1H NMR spectroscopy.
| Entry | Catalyst | % mol | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|---|
| 1 | MgCl2 · 6H2O | 9.5 | – | – | – | – |
| 2 | CuCl2 · 2H2O | 9.5 | 100 | – | – | – |
| 3 | ZnCl2 | 9.5 | 80 | – | – | 20 |
| 4 | AlCl3 | 5 | – | – | – | 100 |
| 5 | BiCl3 | 9.5 | 50 | 40 | 10 | – |
| 6 | BiI3 | 9.5 | 44 | 42 | 12 | 2 |
| 7 | BiPO4 | 9.5 | 53 | 45 | – | 2 |
| 8 | Bi(OTf)3 | 9.5 | 80 | 20 | – | – |
| 9 | Bi(NO3)3 | 0.095 | – | – | – | 100 |
| 10 | Bi(NO3)3 | 0.18 | 40 | – | – | 60 |
| 11 | Bi(NO3)3 | 0.32 | 50 | 50 | – | – |
| 12 | Bi(NO3)3 | 0.65 | 33 | 67 | – | – |
| 13 | Bi(NO3)3 | 0.95 | 90 | 10 | – | – |
| 14 | Bi(NO3)3 | 9.5 | 95 | <5 | – | – |
| 15a | Bi(NO3)3 | 25 | 100 | – | – | – |
aAs reported in [5].
To determine whether the reaction proceeds with other Lewis acids, we explored the use of MgCl2, CuCl2, ZnCl2, AlCl3, BiCl3, BiI3, BiPO4, Bi(OTf)3 and Bi(NO3)3 under the conditions described above. Except for MgCl2, which gave none of the byproducts, all of these Lewis acids catalyzed the reaction to give 1 and/or 4 in amounts ranging from traces to moderate yields. Bismuth salts also produced 3. The results showed that a bismuth salt was necessary to obtain calix[5]pyrrole 2, with the best results being obtained with Bi(NO3)3. The advantages of the method described here—namely that bismuth is relatively non-toxic, the macrocycle is obtained in moderate yield, and the synthesis proceeds without any intermediates—make it the best route to β-unsubstituted meso-decamethylcalix[5]pyrrole reported to date.
Experimental
meso-Decamethylcalix[5]pyrrole (2). In a typical reaction, 6 mg of Bi(NO3)3, 2 mL of acetone and 0.09 mL of pyrrole were mixed with stirring at room temperature for 6 h. The reaction mixture was filtered and the solvent evaporated without heat. Reactants were not distilled prior to use and heat was avoided throughout the process. meso-Decamethylcalix[5]pyrrole was purified from the crude reaction mixture using an Agilent Technologies HPLC 1200 system equipped with a multiple wavelength detector (G1365D) operating at 350 nm. Purification was performed on an analytical Zorbax Eclipse XDB-C18 column (150 × 4.6 mm, Agilent Tech. Santa Clara, CA, USA). The column temperature was maintained at room temperature and the mobile phases consisted of solvent A (80% MeOH/20% H2O) and solvent B (100% EtOAc). Separations were performed by the following solvent gradient: 0 min 20% B, 2.5 min 22.5% B, 20–22.5 min 50% B, 24–26 min 80% B, 31–34 min 100% B, 42–47 min 20% B. All increases of solvent B were linearly programmed. The flow rate was 1 mL/min and the injection volume 20 µL. Yield ca. 25%; mp 208–210 °C; 1H NMR (400 MHz, CDCl3): 1.51 (s, 30H, CH3), 5.77 (d, J = 2.8 Hz, 10H, CH), 7.54 (bs, 5H, NH); 13C NMR: 29.3 (CH3), 35.3 (C(CH3)2), 102.8 (CH), 138.5 (β-C pyrrole); EIMS m/z: 535 (M•+).
References
-
Gale, P. A.; Sessler, J. L.; Král, V. Chem. Commun. 1998, 1–8. doi:10.1039/a706280j
Return to citation in text: [1] -
Gale, P. A.; Anzenbacher, P., Jr.; Sessler, J. L. Coord. Chem. Rev. 2001, 222, 57–102. doi:10.1016/S0010-8545(01)00346-0
Return to citation in text: [1] [2] -
Radha Kishan, M.; Radha Rani, V.; Kulkarni, S. J.; Raghavan, K. V. J. Mol. Catal. A: Chem. 2005, 237, 155–160. doi:10.1016/j.molcata.2005.03.006
Return to citation in text: [1] -
Dey, S.; Pal, K.; Sarkar, S. Tetrahedron Lett. 2006, 47, 5851–5854. doi:10.1016/j.tetlet.2006.06.085
Return to citation in text: [1] -
Mejía-Farfán, I.; Contreras-Celedon, C.; Avina-Verduzco, J.; Chacón-García, L. Lett. Org. Chem. 2008, 5, 237–239. doi:10.2174/157017808783955808
Return to citation in text: [1] [2] [3] [4] -
Turner, B.; Botoshansky, M.; Eichen, Y. Angew. Chem. 1998, 110, 2633–2637. doi:10.1002/(SICI)1521-3757(19980918)110:18<2633::AID-ANGE2633>3.0.CO;2-7
Angew. Chem., Int. Ed. 1998, 37, 2475–2478. doi:10.1002/(SICI)1521-3773(19981002)37:18<2475::AID-ANIE2475>3.0.CO;2-7
Return to citation in text: [1] [2] -
Cafeo, G.; Kohnke, F. H.; La Torre, G. L.; White, A. J. P.; Williams, D. J. Angew. Chem. 2000, 112, 1556–1558. doi:10.1002/(SICI)1521-3757(20000417)112:8<1556::AID-ANGE1556>3.0.CO;2-B
Angew. Chem., Int. Ed. 2000, 39, 1496–1498. doi:10.1002/(SICI)1521-3773(20000417)39:8<1496::AID-ANIE1496>3.0.CO;2-I
Return to citation in text: [1] -
Cafeo, G.; Kohnke, F. H.; Parisi, M. F.; Pistone Nascone, R.; La Torre, G. L.; Williams, D. J. Org. Lett. 2002, 4, 2695–2697. doi:10.1021/ol0262082
Return to citation in text: [1] [2] [3] -
Gale, P. A.; Genge, J. W.; Král, V.; McKervey, M. A.; Sessler, J. L.; Walker, A. Tetrahedron Lett. 1997, 38, 8443–8444. doi:10.1016/S0040-4039(97)10275-1
Return to citation in text: [1] -
Sessler, J. L.; Anzenbacher, P., Jr.; Shriver, J. A.; Jursíková, K.; Lynch, V. M.; Marquez, M. J. Am. Chem. Soc. 2000, 122, 12061–12062. doi:10.1021/ja005650h
Return to citation in text: [1]
| 1. | Gale, P. A.; Sessler, J. L.; Král, V. Chem. Commun. 1998, 1–8. doi:10.1039/a706280j |
| 2. | Gale, P. A.; Anzenbacher, P., Jr.; Sessler, J. L. Coord. Chem. Rev. 2001, 222, 57–102. doi:10.1016/S0010-8545(01)00346-0 |
| 8. | Cafeo, G.; Kohnke, F. H.; Parisi, M. F.; Pistone Nascone, R.; La Torre, G. L.; Williams, D. J. Org. Lett. 2002, 4, 2695–2697. doi:10.1021/ol0262082 |
| 10. | Sessler, J. L.; Anzenbacher, P., Jr.; Shriver, J. A.; Jursíková, K.; Lynch, V. M.; Marquez, M. J. Am. Chem. Soc. 2000, 122, 12061–12062. doi:10.1021/ja005650h |
| 8. | Cafeo, G.; Kohnke, F. H.; Parisi, M. F.; Pistone Nascone, R.; La Torre, G. L.; Williams, D. J. Org. Lett. 2002, 4, 2695–2697. doi:10.1021/ol0262082 |
| 9. | Gale, P. A.; Genge, J. W.; Král, V.; McKervey, M. A.; Sessler, J. L.; Walker, A. Tetrahedron Lett. 1997, 38, 8443–8444. doi:10.1016/S0040-4039(97)10275-1 |
| 6. |
Turner, B.; Botoshansky, M.; Eichen, Y. Angew. Chem. 1998, 110, 2633–2637. doi:10.1002/(SICI)1521-3757(19980918)110:18<2633::AID-ANGE2633>3.0.CO;2-7
Angew. Chem., Int. Ed. 1998, 37, 2475–2478. doi:10.1002/(SICI)1521-3773(19981002)37:18<2475::AID-ANIE2475>3.0.CO;2-7 |
| 7. |
Cafeo, G.; Kohnke, F. H.; La Torre, G. L.; White, A. J. P.; Williams, D. J. Angew. Chem. 2000, 112, 1556–1558. doi:10.1002/(SICI)1521-3757(20000417)112:8<1556::AID-ANGE1556>3.0.CO;2-B
Angew. Chem., Int. Ed. 2000, 39, 1496–1498. doi:10.1002/(SICI)1521-3773(20000417)39:8<1496::AID-ANIE1496>3.0.CO;2-I |
| 2. | Gale, P. A.; Anzenbacher, P., Jr.; Sessler, J. L. Coord. Chem. Rev. 2001, 222, 57–102. doi:10.1016/S0010-8545(01)00346-0 |
| 3. | Radha Kishan, M.; Radha Rani, V.; Kulkarni, S. J.; Raghavan, K. V. J. Mol. Catal. A: Chem. 2005, 237, 155–160. doi:10.1016/j.molcata.2005.03.006 |
| 4. | Dey, S.; Pal, K.; Sarkar, S. Tetrahedron Lett. 2006, 47, 5851–5854. doi:10.1016/j.tetlet.2006.06.085 |
| 5. | Mejía-Farfán, I.; Contreras-Celedon, C.; Avina-Verduzco, J.; Chacón-García, L. Lett. Org. Chem. 2008, 5, 237–239. doi:10.2174/157017808783955808 |
| 5. | Mejía-Farfán, I.; Contreras-Celedon, C.; Avina-Verduzco, J.; Chacón-García, L. Lett. Org. Chem. 2008, 5, 237–239. doi:10.2174/157017808783955808 |
| 5. | Mejía-Farfán, I.; Contreras-Celedon, C.; Avina-Verduzco, J.; Chacón-García, L. Lett. Org. Chem. 2008, 5, 237–239. doi:10.2174/157017808783955808 |
| 6. |
Turner, B.; Botoshansky, M.; Eichen, Y. Angew. Chem. 1998, 110, 2633–2637. doi:10.1002/(SICI)1521-3757(19980918)110:18<2633::AID-ANGE2633>3.0.CO;2-7
Angew. Chem., Int. Ed. 1998, 37, 2475–2478. doi:10.1002/(SICI)1521-3773(19981002)37:18<2475::AID-ANIE2475>3.0.CO;2-7 |
| 8. | Cafeo, G.; Kohnke, F. H.; Parisi, M. F.; Pistone Nascone, R.; La Torre, G. L.; Williams, D. J. Org. Lett. 2002, 4, 2695–2697. doi:10.1021/ol0262082 |
| 5. | Mejía-Farfán, I.; Contreras-Celedon, C.; Avina-Verduzco, J.; Chacón-García, L. Lett. Org. Chem. 2008, 5, 237–239. doi:10.2174/157017808783955808 |
© 2009 Chacón-García et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)