Abstract
Using a combination of commercially available mesofluidic flow equipment and tubes packed with immobilised reagents and scavengers, a new synthesis of α-ketoesters is reported.

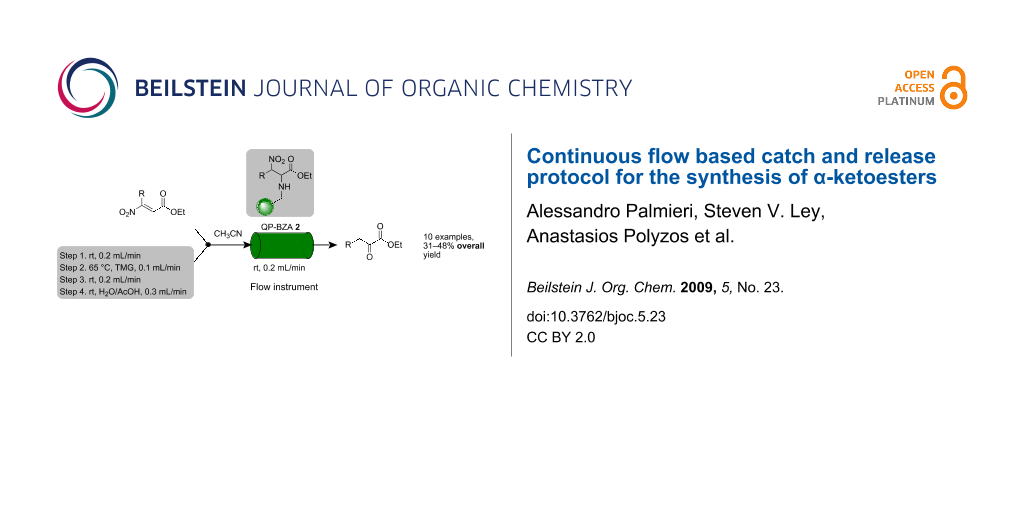
Graphical Abstract
Introduction
Organic synthesis is changing rapidly owing to the discovery of processes that challenge current dogma and lead to the invention of new chemical reactions [1,2]. Likewise, new synthesis tools are impacting on the way we assemble molecules. Of these, flow chemistry technologies are becoming especially important [3-14]. For many years, our group [15-22] has been focussed on using immobilised systems [23-29] to more effectively and cleanly bring about chemical transformations, especially in multistep mode [17,30-37]. Given the success of these concepts, it is not surprising that we would want to adapt these principles to various flow-chemical synthesis platforms to effectuate automated multistep chemical syntheses [38-54].
In this work we report the use of the Uniqsis FlowSyn™ continuous flow reactor [55] (Figure 1) to effect a flow-based preparation of α-ketoesters. The key feature of this process is the application of a catch and release protocol [56-72], under the flow reaction conditions. Our choice of α-ketoesters as products of the process was governed by their use as starting materials for various synthesis programmes [73-81] and as important products in their own right [82-88]. Common methods for the preparation of α-ketoesters include the modified Dakin-West reaction [89] and the addition of a Grignard reagent to oxalates or oxalyl chlorides [90-92] together with a few alternative syntheses [93-99]. These procedures often suffer from drastic conditions, restricted selectivity and poor yields. Our flow-based approach delivers a new and general method for the preparation of α-ketoesters, which proceeds under mild conditions, with good functional group tolerability and generates products in high purity.
![[1860-5397-5-23-1]](/bjoc/content/figures/1860-5397-5-23-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The Uniqsis FlowSyn™ continuous flow reactor comprising of a column holder and heating unit (A) and the reactor coil (B). A detailed image of the reactor coil is shown on the right.
Figure 1: The Uniqsis FlowSyn™ continuous flow reactor comprising of a column holder and heating unit (A) and...
Results and Discussion
The experimental set up for these transformations involves the use of the Uniqsis FlowSyn™ device [55]. The fully integrated instrument employs a dual channel flow system, with each channel independently driven by a variable high-pressure piston pump. The starting materials and reagents are dispensed from sample loops (0.5–10 mL) and are united in a T-mixing piece and then passed into either a coil or column reactor (Figure 1). The column reactor utilises adjustable glass columns with variable internal diameter (1–1.5 cm) and range in volume from 6–83 mL (unpacked). The coil reactors are made from a selection of materials including PTFE, PEEK, stainless steel or Hastelloy® and accommodate volumes from 2–20 mL. The column reactor (Figure 1, A) can be heated up to 150 °C and the coil heater (Figure 1, B) up to 260 °C, over a range of flow rates between 0.01–20 mL/min, and can be configured for multistep or parallel operation. Exiting products can be collected as aliquots using an automated fraction collector for reaction optimisation or as a bulk sample for scale-up. In addition, product purification can be achieved as part of the overall flow process by in-line solid phase extraction (SPE) or alternatively by diverting the flow stream into an attached HPLC system [100].
A series of preliminary experiments was carried out on the flow equipment to profile the reaction in terms of optimum reaction temperature, concentration, residence time, solvent and stoichiometry. Following rapid screening of conditions, we fixed upon a set of reaction parameters for efficient synthesis of α-ketoesters (Scheme 1). The overall reaction process proceeds in the flow apparatus via nitroolefinic esters 1 as substrates which are captured onto a benzylamine polymer 2 (QuadraPure™ QP-BZA polymer, loading 5.5 mmol/g, 4 equiv) to give 3 to effect product clean-up. In this way the immobilised species 3 can be washed and any solution phase impurities (resulting from the formation of the nitroolefinic ester – see later) are directed to waste (step 1). Next the column is treated with a flow stream of tetramethylguanidine (TMG; step 2) to cause elimination of nitrous acid and produce the corresponding enamino acid esters, which remain attached to the polymer support. Finally, after flow-washing (step 3), the solid supported species is hydrolysed, liberating α-ketoester product 4 by flowing aqueous acetic acid (step 4) through the in-line column. The overall route constitutes a new flow chemistry example of the catch-react-and-release concept that we have used successfully in other synthesis programmes [101-103].
![[1860-5397-5-23-i1]](/bjoc/content/inline/1860-5397-5-23-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General procedure for the flow synthesis of α-ketoester products 4a–j.
Scheme 1: General procedure for the flow synthesis of α-ketoester products 4a–j.
The nitroolefinic esters 1 were originally formed in a separate batch reaction from a Henry coupling of appropriate nitro compounds with ethyl glyoxalate over Amberlyst™ 21 (A21) resin to give the corresponding nitroalkanol 5 [104]. This was followed by treatment of 5 with methanesulfonyl chloride (MsCl) or trifluoroacetic anhydride (TFAA) to promote the base-catalysed dehydration, affording the nitroolefinic esters 1 (Scheme 2) [105]. As we have deliberately constructed this sequence for implementation in a continuous flow process, the intermediate nitroalkanols 5 were not isolated and the nitroolefinic esters were used without further purification. The average yield for the nitroolefins 1a–j prepared as described in Scheme 2 was approximately 60% by LCMS. Impurities were readily removed following immobilisation of nitroolefinic esters 1 on the QP-BZA resin.
![[1860-5397-5-23-i2]](/bjoc/content/inline/1860-5397-5-23-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General procedure for the batch synthesis of nitroolefinic esters 1a–j.
Scheme 2: General procedure for the batch synthesis of nitroolefinic esters 1a–j.
In addition, the flow synthesis of two representative compounds was undertaken to allow for the complete generation of α-ketoester products in flow from the starting nitroalkanes (Scheme 3). As shown in Table 1, we demonstrate that the synthesis of the nitroolefinic esters was achieved under flow conditions in a clean and effective fashion. Moreover, this synthesis demonstrates the first reported example of Henry reaction conducted in flow and we intend to elaborate on this important transformation in future studies.
![[1860-5397-5-23-i3]](/bjoc/content/inline/1860-5397-5-23-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: General procedure for the flow synthesis of nitroolefinic esters 1a,c.
Scheme 3: General procedure for the flow synthesis of nitroolefinic esters 1a,c.
Table 1: Nitroolefinic esters 1a,c prepared under flow conditions (as described in Scheme 3).
| Entry | 1 | Compound | Yield (%)a |
|---|---|---|---|
| 1 | 1a |
![[Graphic 1]](/bjoc/content/inline/1860-5397-5-23-i4.svg?max-width=637&scale=1.0)
|
63 |
| 2 | 1c |
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-23-i5.svg?max-width=637&scale=1.0)
|
55 |
aIsolated yields are shown.
Figure 2 illustrates the examples and yields of α-ketoester products afforded by this new approach. While the list is not extensive, we have established that the process is tolerant of both aliphatic and aromatic substituted nitro-derivatives in the first step, and accommodates ester, acetate, acetal, nitrile and olefinic functionality in the final product. The process was reliable over several runs and consistently afforded very clean material (≥ 97% by NMR). The yields while only moderate for the overall process still equate to an average step conversion of 68–78% per chemical iteration, given that the sequence is a multistep process (see Supporting Information for full experimental data).
![[1860-5397-5-23-2]](/bjoc/content/figures/1860-5397-5-23-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: α-Ketoesters prepared and isolated yields.
Figure 2: α-Ketoesters prepared and isolated yields.
Conclusion
In conclusion, we have demonstrated the versatility of the Uniqsis FlowSyn™ unit to achieve multi-step organic synthesis under continuous flow-chemistry conditions. This was accomplished by adapting the device to incorporate immobilised reagents packed in flow tubes, enabling clean transformations without recourse to conventional product work-up or purification. The overall process delivers synthetically useful α-ketoester products in high purity from various nitroalkane inputs and paves the way for more extended reaction sequences.
Supporting Information
| Supporting Information File 1: Supporting Information – Continuous flow based catch and release protocol for the synthesis of α-ketoesters | ||
| Format: DOC | Size: 45.5 KB | Download |
References
-
Ley, S. V.; Baxendale, I. R. Nat. Rev. Drug Discovery 2002, 1, 573–586. doi:10.1038/nrd871
Return to citation in text: [1] -
Baxendale, I. R.; Hayward, J. J.; Ley, S. V.; Tranmer, G. K. ChemMedChem 2007, 2, 768–788. doi:10.1002/cmdc.200700008
Return to citation in text: [1] -
Baxendale, I. R.; Pitts, M. R. Chim. Oggi 2006, 24 (3), 41–45.
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V. Heterogeneous Reactions. In New Avenues to Efficient Chemical Synthesis, Emerging Technologies; Seeberger, P. H.; Blume, T., Eds.; Springer-Verlag: Berlin, Heidelberg, 2007; pp 151–185.
Return to citation in text: [1] -
Baxendale, I. R.; Hayward, J. J.; Ley, S. V. Comb. Chem. High Throughput Screening 2007, 10, 802–836. doi:10.2174/138620707783220374
Return to citation in text: [1] -
Baxendale, I. R.; Hayward, J. J.; Lanners, S.; Ley, S. V.; Smith, C. D. Heterogeneous Reactions. In Microreactors in Organic Synthesis and Catalysis; Wirth, T., Ed.; Wiley-VCH: Weinheim, 2008; pp 84–122.
Chapter 4.2.
Return to citation in text: [1] -
Jas, G.; Kirschning, A. Chem.–Eur. J. 2003, 9, 5708–5723. doi:10.1002/chem.200305212
Return to citation in text: [1] -
Hodge, P. Curr. Opin. Chem. Biol. 2003, 7, 362–373. doi:10.1016/S1367-5931(03)00052-8
Return to citation in text: [1] -
Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem. 2004, 116, 410–451. doi:10.1002/ange.200300577
Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577
Return to citation in text: [1] -
Kirschning, A.; Solodenko, W.; Mennecke, K. Chem.–Eur. J. 2006, 12, 5972–5990. doi:10.1002/chem.200600236
Return to citation in text: [1] -
Ahmed-Omer, B.; Brandt, J. C.; Wirth, T. Org. Biomol. Chem. 2007, 5, 733–740. doi:10.1039/b615072a
Return to citation in text: [1] -
Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c
Return to citation in text: [1] -
Glasnov, V. T. N.; Kappe, C. O. Macromol. Rapid Commun. 2007, 28, 395–410. doi:10.1002/marc.200600665
Return to citation in text: [1] -
Benito-López, F.; Egberink, R. J. M.; Reinhoudt, D. N.; Verboom, W. Tetrahedron 2008, 64, 10023–10040. doi:10.1016/j.tet.2008.07.108
Return to citation in text: [1] -
Ley, S. V.; Baxendale, I. R.; Bream, R. N.; Jackson, P. S.; Leach, A. G.; Longbottom, D. A.; Nesi, M.; Scott, J. S.; Storer, R. I.; Taylor, S. J. J. Chem. Soc., Perkin Trans. 1 2000, 3815–4195. doi:10.1039/b006588i
Return to citation in text: [1] -
Baxendale, I. R.; Lee, A.-L.; Ley, S. V. J. Chem. Soc., Perkin Trans. 1 2002, 1850–1857. doi:10.1039/b203388g
Return to citation in text: [1] -
Baxendale, I. R.; Ernst, M.; Krahnert, W.-R.; Ley, S. V. Synlett 2002, 1641–1644. doi:10.1055/s-2002-34249
Return to citation in text: [1] [2] -
Ley, S. V.; Baxendale, I. R. Chem. Rec. 2002, 2, 377–388. doi:10.1002/tcr.10033
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V.; Nesi, M.; Piutti, C. Tetrahedron 2002, 58, 6285–6304. doi:10.1016/S0040-4020(02)00628-2
Return to citation in text: [1] -
Storer, R. I.; Takemoto, T.; Jackson, P. S.; Ley, S. V. Angew. Chem. 2003, 115, 2625–2629. doi:10.1002/ange.200351413
Angew. Chem., Int. Ed. 2003, 42, 2521–2525. doi:10.1002/anie.200351413
Return to citation in text: [1] -
Storer, R. I.; Takemoto, T.; Jackson, P. S.; Brown, D. S.; Baxendale, I. R.; Ley, S. V. Chem.–Eur. J. 2004, 10, 2529–2547. doi:10.1002/chem.200305669
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V. Ind. Eng. Chem. Res. 2005, 44, 8588–8592. doi:10.1021/ie048822i
Return to citation in text: [1] -
Thompson, L. A. Curr. Opin. Chem. Biol. 2000, 4, 324–337. doi:10.1016/S1367-5931(00)00096-X
Return to citation in text: [1] -
Kobayashi, S. Curr. Opin. Chem. Biol. 2000, 4, 338–345. doi:10.1016/S1367-5931(00)00097-1
Return to citation in text: [1] -
Kirschning, A.; Monenschein, H.; Wittenberg, R. Chem.–Eur. J. 2000, 6, 4445–4450. doi:10.1002/1521-3765(20001215)6:24<4445::AID-CHEM4445>3.0.CO;2-W
Return to citation in text: [1] -
Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem. 2001, 113, 670–701. doi:10.1002/1521-3757(20010216)113:4<670::AID-ANGE6700>3.0.CO;2-G
Angew. Chem., Int. Ed. 2001, 40, 650–679. doi:10.1002/1521-3773(20010216)40:4<650::AID-ANIE6500>3.0.CO;2-C
Return to citation in text: [1] -
Sherrington, D. C. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2364–2377. doi:10.1002/pola.1213
Return to citation in text: [1] -
Hodge, P. Ind. Eng. Chem. Res. 2005, 44, 8542–8553. doi:10.1021/ie040285e
Return to citation in text: [1] -
Solinas, A.; Taddei, M. Synthesis 2007, 2409–2453. doi:10.1055/s-2007-983806
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V. Bioorg. Med. Chem. Lett. 2000, 10, 1983–1986. doi:10.1016/S0960-894X(00)00383-8
Return to citation in text: [1] -
Ley, S. V.; Baxendale, I. R.; Brusotti, G.; Caldarelli, M.; Massi, A.; Nesi, M. Farmaco 2002, 57, 321–330. doi:10.1016/S0014-827X(02)01210-7
Return to citation in text: [1] -
Baxendale, I. R.; Brusotti, G.; Matsuoka, M.; Ley, S. V. J. Chem. Soc., Perkin Trans. 1 2002, 143–154. doi:10.1039/b109482n
Return to citation in text: [1] -
Baxendale, I. R.; Lee, A.-L.; Ley, S. V. Synlett 2002, 516–518. doi:10.1055/s-2002-20483
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V.; Lumeras, W.; Nesi, M. Comb. Chem. High Throughput Screening 2002, 5, 197–199.
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V.; Sneddon, H. F. Synlett 2002, 775–777. doi:10.1055/s-2002-25333
Return to citation in text: [1] -
Baxendale, I. R.; Storer, R. I.; Ley, S. V. Supported Reagents and Scavengers in Multi-Step Organic Synthesis. In Polymeric Materials in Organic Synthesis and Catalysis; Buchmeiser, M. R., Ed.; Wiley-VCH: Weinheim, 2003; pp 53–136. doi:10.1002/3527601856.ch2
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V. Curr. Org. Chem. 2005, 9, 1521–1534. doi:10.2174/138527205774370513
Return to citation in text: [1] -
Baxendale, I. R.; Deeley, J.; Griffiths-Jones, C. M.; Ley, S. V.; Saaby, S.; Tranmer, G. K. Chem. Commun. 2006, 2566–2568. doi:10.1039/b600382f
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Org. Lett. 2006, 8, 5231–5234. doi:10.1021/ol061975c
Return to citation in text: [1] -
Smith, C. J.; Iglesias-Sigüenza, F. J.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 2758–2761. doi:10.1039/b709043a
Return to citation in text: [1] -
Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k
Return to citation in text: [1] -
Hornung, C. H.; Mackley, M. R.; Baxendale, I. R.; Ley, S. V. Org. Process Res. Dev. 2007, 11, 399–405. doi:10.1021/op700015f
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V. Synlett 2008, 2111–2114. doi:10.1055/s-2008-1078026
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N.; Smith, C. D. Org. Biomol. Chem. 2008, 6, 1587–1593. doi:10.1039/b801634h
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N.; Smith, C. D.; Tierney, J. P. Org. Biomol. Chem. 2008, 6, 1577–1586. doi:10.1039/b801631n
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tamborini, L.; Voica, A.-F. J. Comb. Chem. 2008, 10, 851–857. doi:10.1021/cc800070a
Return to citation in text: [1] -
Jas, G.; Kirschning, A. Chem.–Eur. J. 2003, 9, 5708–5723. doi:10.1002/chem.200305212
Return to citation in text: [1] -
Bernstein, D.; France, S.; Wolfer, J.; Lectka, T. Tetrahedron: Asymmetry 2005, 16, 3481–3483. doi:10.1016/j.tetasy.2005.09.014
Return to citation in text: [1] -
Bonfils, F.; Cazaux, I.; Hodge, P.; Caze, C. Org. Biomol. Chem. 2006, 4, 493–497. doi:10.1039/b515241k
Return to citation in text: [1] -
Wiles, C.; Watts, P.; Haswell, S. J. Tetrahedron Lett. 2006, 47, 5261–5264. doi:10.1016/j.tetlet.2006.05.157
Return to citation in text: [1] -
Dräger, G.; Kiss, C.; Kunz, U.; Kirschning, A. Org. Biomol. Chem. 2007, 5, 3657–3664. doi:10.1039/b712804e
Return to citation in text: [1] -
Burguete, M. I.; Cornejo, A.; García-Verdugo, E.; Gil, M. J.; Luis, S. V.; Mayoral, J. A.; Martínez-Merino, V.; Sokolova, M. J. Org. Chem. 2007, 72, 4344–4350. doi:10.1021/jo070119r
Return to citation in text: [1] -
Solodenko, W.; Jas, G.; Kunz, U.; Kirschning, A. Synthesis 2007, 583–589. doi:10.1055/s-2007-965877
Return to citation in text: [1] -
Odedra, A.; Geyer, K.; Gustafsson, T.; Gilmour, R.; Seeberger, P. H. Chem. Commun. 2008, 3025–3027. doi:10.1039/b803715a
Return to citation in text: [1] -
Uniqsis web site. http://www.uniqsis.com (accessed April 6, 2009).
Return to citation in text: [1] [2] -
Cohen, B. J.; Kraus, M. A.; Patchornik, A. J. Am. Chem. Soc. 1977, 99, 4165–4167. doi:10.1021/ja00454a050
Return to citation in text: [1] -
Cohen, B. J.; Kraus, M. A.; Patchornik, A. J. Am. Chem. Soc. 1981, 103, 7620–7629. doi:10.1021/ja00415a034
Return to citation in text: [1] -
Patchornik, A. In Proc. IUPAC, I. U. P. A. C., Macromol. Symp., 28th, 1982, University of Massachusetts, Amherst, July 12–16, 1982; IUPAC: Oxford, 1982; p 85.
Return to citation in text: [1] -
Brown, S. D.; Armstrong, R. W. J. Am. Chem. Soc. 1996, 118, 6331–6332. doi:10.1021/ja961203j
Return to citation in text: [1] -
Hu, Y.; Baudart, S.; Porco, J. A., Jr. J. Org. Chem. 1999, 64, 1049–1051. doi:10.1021/jo981874v
Return to citation in text: [1] -
Studer, A.; Hadida, S.; Ferritto, R.; Kim, S.-Y.; Jeger, P.; Wipf, P.; Curran, D. P. Science 1997, 275, 823–826. doi:10.1126/science.275.5301.823
Return to citation in text: [1] -
Flynn, D. L.; Crich, J. Z.; Devraj, R. V.; Hockerman, S. L.; Parlow, J. J.; South, M. S.; Woodard, S. J. Am. Chem. Soc. 1997, 119, 4874–4881. doi:10.1021/ja963462e
Return to citation in text: [1] -
Curran, D. P. Angew. Chem. 1998, 110, 1230–1255. doi:10.1002/(SICI)1521-3757(19980504)110:9<1230::AID-ANGE1230>3.0.CO;2-Y
Angew. Chem., Int. Ed. 1998, 37, 1174–1196. doi:10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P
Return to citation in text: [1] -
Bosanac, T.; Yang, J.; Wilcox, C. S. Angew. Chem. 2001, 113, 1927–1931. doi:10.1002/1521-3757(20010518)113:10<1927::AID-ANGE1927>3.0.CO;2-#
Angew. Chem., Int. Ed. 2001, 40, 1875–1879. doi:10.1002/1521-3773(20010518)40:10<1875::AID-ANIE1875>3.0.CO;2-5
Return to citation in text: [1] -
Ley, S. V.; Massi, A.; Rodríguez, F.; Horwell, D. C.; Lewthwaite, R. A.; Pritchard, M. C.; Reid, A. M. Angew. Chem. 2001, 113, 1088–1090. doi:10.1002/1521-3757(20010316)113:6<1088::AID-ANGE10880>3.0.CO;2-#
Angew. Chem., Int. Ed. 2001, 40, 1053–1055. doi:10.1002/1521-3773(20010316)40:6<1053::AID-ANIE10530>3.0.CO;2-D
Return to citation in text: [1] -
Galante, A.; Lhoste, P.; Sinou, D. Tetrahedron Lett. 2001, 42, 5425–5427. doi:10.1016/S0040-4039(01)01055-3
Return to citation in text: [1] -
Yoshida, J.-i.; Itami, K. Chem. Rev. 2002, 102, 3693–3716. doi:10.1021/cr0103524
Return to citation in text: [1] -
Dobbs, A. P.; McGregor-Johnson, C. Tetrahedron Lett. 2002, 43, 2807–2810. doi:10.1016/S0040-4039(02)00322-2
Return to citation in text: [1] -
Lan, P.; Porco, J. A., Jr.; South, M. S.; Parlow, J. J. J. Comb. Chem. 2003, 5, 660–669. doi:10.1021/cc030028h
Return to citation in text: [1] -
Siu, J.; Baxendale, I. R.; Lewthwaite, R. A.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3140–3160. doi:10.1039/b503778f
Return to citation in text: [1] -
Curran, D. P.; Wang, X.; Zhang, Q. J. Org. Chem. 2005, 70, 3716–3719. doi:10.1021/jo050116j
Return to citation in text: [1] -
Mothana, S.; Chahal, N.; Vanneste, S.; Hall, D. G. J. Comb. Chem. 2007, 9, 193–196. doi:10.1021/cc060149s
Return to citation in text: [1] -
Audrain, H.; Thorhauge, J.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2000, 65, 4487–4497. doi:10.1021/jo9918596
Return to citation in text: [1] -
Peng, Z.-H.; Woerpel, K. A. Org. Lett. 2002, 4, 2945–2948. doi:10.1021/ol026343e
Return to citation in text: [1] -
Yu, S.; Saenz, J.; Srirangam, J. K. J. Org. Chem. 2002, 67, 1699–1702. doi:10.1021/jo016131f
Return to citation in text: [1] -
Griesbeck, A. G.; Bondock, S.; Lex, J. Org. Biomol. Chem. 2004, 2, 1113–1115. doi:10.1039/b401990c
Return to citation in text: [1] -
Sun, Y.; Wan, X.; Wang, J.; Meng, Q.; Zhang, H.; Jiang, L.; Zhang, Z. Org. Lett. 2005, 7, 5425–5427. doi:10.1021/ol052212c
Return to citation in text: [1] -
Zhang, W.; Shi, M. Chem. Commun. 2006, 1218–1220. doi:10.1039/b516467b
Return to citation in text: [1] -
Howard, B. E.; Woerpel, K. A. Org. Lett. 2007, 9, 4651–4653. doi:10.1021/ol702148x
Return to citation in text: [1] -
Kratzer, R.; Nidetzky, B. Chem. Commun. 2007, 1047–1049. doi:10.1039/b616475g
Return to citation in text: [1] -
Ntaganda, R.; Milovic, T.; Tiburcio, J.; Thadani, A. N. Chem. Commun. 2008, 4052–4054. doi:10.1039/b808302a
Return to citation in text: [1] -
Peet, N. P.; Burkhart, J. P.; Angelastro, M. R.; Giroux, E. L.; Mehdi, S.; Bey, P.; Kolb, M.; Neises, B.; Schirlin, D. J. Med. Chem. 1990, 33, 394–407. doi:10.1021/jm00163a063
Return to citation in text: [1] -
Patel, D. V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C. A.; Smith, S. A.; Petrillo, E. W., Jr. J. Med. Chem. 1993, 36, 2431–2447. doi:10.1021/jm00069a001
Return to citation in text: [1] -
Li, Z.; Patil, G. S.; Golubski, Z. E.; Hori, H.; Tehrani, K.; Foreman, J. E.; Eveleth, D. D.; Bartus, R. T.; Powers, J. C. J. Med. Chem. 1993, 36, 3472–3480. doi:10.1021/jm00074a031
Return to citation in text: [1] -
Koutek, B.; Prestwich, G. D.; Howlett, A. C.; Chin, S. A.; Salehani, D.; Akhavan, N.; Deutsch, D. G. J. Biol. Chem. 1994, 269, 22937–22940.
Return to citation in text: [1] -
Conde-Frieboes, K.; Reynolds, L. J.; Lio, Y.-C.; Hale, M. R.; Wasserman, H. H.; Dennis, E. A. J. Am. Chem. Soc. 1996, 118, 5519–5525. doi:10.1021/ja953553w
Return to citation in text: [1] -
Otto, H.-H.; Schirmeister, T. Chem. Rev. 1997, 97, 133–172. doi:10.1021/cr950025u
Return to citation in text: [1] -
Choe, Y.; Brinen, L. S.; Price, M. S.; Engel, J. C.; Lange, M.; Grisostomi, C.; Weston, S. G.; Pallai, P. V.; Cheng, H.; Hardy, L. W.; Hartsough, D. S.; McMakin, M.; Tilton, R. F.; Baldino, C. M.; Craik, C. S. Bioorg. Med. Chem. 2005, 13, 2141–2156. doi:10.1016/j.bmc.2004.12.053
Return to citation in text: [1] -
Buchanan, G. L. Chem. Soc. Rev. 1988, 17, 91–109. doi:10.1039/cs9881700091
Return to citation in text: [1] -
Nimitz, J. S.; Mosher, H. S. J. Org. Chem. 1981, 46, 211–213. doi:10.1021/jo00314a057
Return to citation in text: [1] -
Creary, X.; Mehrsheikh-Mohammadi, M. E. J. Org. Chem. 1986, 51, 2664–2668. doi:10.1021/jo00364a009
Return to citation in text: [1] -
Babudri, F.; Fiandanese, V.; Marchese, G.; Punzi, A. Tetrahedron 1996, 52, 13513–13520. doi:10.1016/0040-4020(96)00805-8
Return to citation in text: [1] -
Wasserman, H. H.; Ives, J. L. J. Org. Chem. 1985, 50, 3573–3580. doi:10.1021/jo00219a025
Return to citation in text: [1] -
Bulman Page, P. C.; Rosenthal, S. Tetrahedron Lett. 1986, 27, 1947–1950. doi:10.1016/S0040-4039(00)84419-6
Return to citation in text: [1] -
Sakakura, T.; Yamashita, H.; Kobayashi, T.-a.; Hayashi, T.; Tanaka, M. J. Org. Chem. 1987, 52, 5733–5740. doi:10.1021/jo00235a017
Return to citation in text: [1] -
Wong, M.-K.; Yu, C.-W.; Yuen, W.-H.; Yang, D. J. Org. Chem. 2001, 66, 3606–3609. doi:10.1021/jo0015974
Return to citation in text: [1] -
Li, L.-S.; Wu, Y.-L. Tetrahedron Lett. 2002, 43, 2427–2430. doi:10.1016/S0040-4039(02)00290-3
Return to citation in text: [1] -
Ma, M.; Li, C.; Peng, L.; Xie, F.; Zhang, X.; Wang, J. Tetrahedron Lett. 2005, 46, 3927–3929. doi:10.1016/j.tetlet.2005.03.199
Return to citation in text: [1] -
Shimizu, H.; Murakami, M. Chem. Commun. 2007, 2855–2857. doi:10.1039/b704105e
Return to citation in text: [1] -
Baxendale, I. R.; Griffiths-Jones, C. M.; Ley, S. V.; Tranmer, G. K. Synlett 2006, 427–430. doi:10.1055/s-2006-926244
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Chem. Commun. 2006, 4835–4837. doi:10.1039/b612197g
Return to citation in text: [1] -
Smith, C. D.; Baxendale, I. R.; Tranmer, G. K.; Baumann, M.; Smith, S. C.; Lewthwaite, R. A.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1562–1568. doi:10.1039/b703033a
Return to citation in text: [1] -
Griffiths-Jones, C. M.; Hopkin, M. D.; Jönsson, D.; Ley, S. V.; Tapolczay, D. J.; Vickerstaffe, E.; Ladlow, M. J. Comb. Chem. 2007, 9, 422–430. doi:10.1021/cc060152b
Return to citation in text: [1] -
Ballini, R.; Bosica, G.; Forconi, P. Tetrahedron 1996, 52, 1677–1684. doi:10.1016/0040-4020(95)00996-5
Return to citation in text: [1] -
Ballini, R.; Fiorini, D.; Palmieri, A. Tetrahedron Lett. 2004, 45, 7027–7029. doi:10.1016/j.tetlet.2004.07.141
Return to citation in text: [1]
| 104. | Ballini, R.; Bosica, G.; Forconi, P. Tetrahedron 1996, 52, 1677–1684. doi:10.1016/0040-4020(95)00996-5 |
| 105. | Ballini, R.; Fiorini, D.; Palmieri, A. Tetrahedron Lett. 2004, 45, 7027–7029. doi:10.1016/j.tetlet.2004.07.141 |
| 1. | Ley, S. V.; Baxendale, I. R. Nat. Rev. Drug Discovery 2002, 1, 573–586. doi:10.1038/nrd871 |
| 2. | Baxendale, I. R.; Hayward, J. J.; Ley, S. V.; Tranmer, G. K. ChemMedChem 2007, 2, 768–788. doi:10.1002/cmdc.200700008 |
| 17. | Baxendale, I. R.; Ernst, M.; Krahnert, W.-R.; Ley, S. V. Synlett 2002, 1641–1644. doi:10.1055/s-2002-34249 |
| 30. | Baxendale, I. R.; Ley, S. V. Bioorg. Med. Chem. Lett. 2000, 10, 1983–1986. doi:10.1016/S0960-894X(00)00383-8 |
| 31. | Ley, S. V.; Baxendale, I. R.; Brusotti, G.; Caldarelli, M.; Massi, A.; Nesi, M. Farmaco 2002, 57, 321–330. doi:10.1016/S0014-827X(02)01210-7 |
| 32. | Baxendale, I. R.; Brusotti, G.; Matsuoka, M.; Ley, S. V. J. Chem. Soc., Perkin Trans. 1 2002, 143–154. doi:10.1039/b109482n |
| 33. | Baxendale, I. R.; Lee, A.-L.; Ley, S. V. Synlett 2002, 516–518. doi:10.1055/s-2002-20483 |
| 34. | Baxendale, I. R.; Ley, S. V.; Lumeras, W.; Nesi, M. Comb. Chem. High Throughput Screening 2002, 5, 197–199. |
| 35. | Baxendale, I. R.; Ley, S. V.; Sneddon, H. F. Synlett 2002, 775–777. doi:10.1055/s-2002-25333 |
| 36. | Baxendale, I. R.; Storer, R. I.; Ley, S. V. Supported Reagents and Scavengers in Multi-Step Organic Synthesis. In Polymeric Materials in Organic Synthesis and Catalysis; Buchmeiser, M. R., Ed.; Wiley-VCH: Weinheim, 2003; pp 53–136. doi:10.1002/3527601856.ch2 |
| 37. | Baxendale, I. R.; Ley, S. V. Curr. Org. Chem. 2005, 9, 1521–1534. doi:10.2174/138527205774370513 |
| 100. | Baxendale, I. R.; Griffiths-Jones, C. M.; Ley, S. V.; Tranmer, G. K. Synlett 2006, 427–430. doi:10.1055/s-2006-926244 |
| 23. | Thompson, L. A. Curr. Opin. Chem. Biol. 2000, 4, 324–337. doi:10.1016/S1367-5931(00)00096-X |
| 24. | Kobayashi, S. Curr. Opin. Chem. Biol. 2000, 4, 338–345. doi:10.1016/S1367-5931(00)00097-1 |
| 25. | Kirschning, A.; Monenschein, H.; Wittenberg, R. Chem.–Eur. J. 2000, 6, 4445–4450. doi:10.1002/1521-3765(20001215)6:24<4445::AID-CHEM4445>3.0.CO;2-W |
| 26. |
Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem. 2001, 113, 670–701. doi:10.1002/1521-3757(20010216)113:4<670::AID-ANGE6700>3.0.CO;2-G
Angew. Chem., Int. Ed. 2001, 40, 650–679. doi:10.1002/1521-3773(20010216)40:4<650::AID-ANIE6500>3.0.CO;2-C |
| 27. | Sherrington, D. C. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2364–2377. doi:10.1002/pola.1213 |
| 28. | Hodge, P. Ind. Eng. Chem. Res. 2005, 44, 8542–8553. doi:10.1021/ie040285e |
| 29. | Solinas, A.; Taddei, M. Synthesis 2007, 2409–2453. doi:10.1055/s-2007-983806 |
| 101. | Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Chem. Commun. 2006, 4835–4837. doi:10.1039/b612197g |
| 102. | Smith, C. D.; Baxendale, I. R.; Tranmer, G. K.; Baumann, M.; Smith, S. C.; Lewthwaite, R. A.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1562–1568. doi:10.1039/b703033a |
| 103. | Griffiths-Jones, C. M.; Hopkin, M. D.; Jönsson, D.; Ley, S. V.; Tapolczay, D. J.; Vickerstaffe, E.; Ladlow, M. J. Comb. Chem. 2007, 9, 422–430. doi:10.1021/cc060152b |
| 15. | Ley, S. V.; Baxendale, I. R.; Bream, R. N.; Jackson, P. S.; Leach, A. G.; Longbottom, D. A.; Nesi, M.; Scott, J. S.; Storer, R. I.; Taylor, S. J. J. Chem. Soc., Perkin Trans. 1 2000, 3815–4195. doi:10.1039/b006588i |
| 16. | Baxendale, I. R.; Lee, A.-L.; Ley, S. V. J. Chem. Soc., Perkin Trans. 1 2002, 1850–1857. doi:10.1039/b203388g |
| 17. | Baxendale, I. R.; Ernst, M.; Krahnert, W.-R.; Ley, S. V. Synlett 2002, 1641–1644. doi:10.1055/s-2002-34249 |
| 18. | Ley, S. V.; Baxendale, I. R. Chem. Rec. 2002, 2, 377–388. doi:10.1002/tcr.10033 |
| 19. | Baxendale, I. R.; Ley, S. V.; Nesi, M.; Piutti, C. Tetrahedron 2002, 58, 6285–6304. doi:10.1016/S0040-4020(02)00628-2 |
| 20. |
Storer, R. I.; Takemoto, T.; Jackson, P. S.; Ley, S. V. Angew. Chem. 2003, 115, 2625–2629. doi:10.1002/ange.200351413
Angew. Chem., Int. Ed. 2003, 42, 2521–2525. doi:10.1002/anie.200351413 |
| 21. | Storer, R. I.; Takemoto, T.; Jackson, P. S.; Brown, D. S.; Baxendale, I. R.; Ley, S. V. Chem.–Eur. J. 2004, 10, 2529–2547. doi:10.1002/chem.200305669 |
| 22. | Baxendale, I. R.; Ley, S. V. Ind. Eng. Chem. Res. 2005, 44, 8588–8592. doi:10.1021/ie048822i |
| 93. | Wasserman, H. H.; Ives, J. L. J. Org. Chem. 1985, 50, 3573–3580. doi:10.1021/jo00219a025 |
| 94. | Bulman Page, P. C.; Rosenthal, S. Tetrahedron Lett. 1986, 27, 1947–1950. doi:10.1016/S0040-4039(00)84419-6 |
| 95. | Sakakura, T.; Yamashita, H.; Kobayashi, T.-a.; Hayashi, T.; Tanaka, M. J. Org. Chem. 1987, 52, 5733–5740. doi:10.1021/jo00235a017 |
| 96. | Wong, M.-K.; Yu, C.-W.; Yuen, W.-H.; Yang, D. J. Org. Chem. 2001, 66, 3606–3609. doi:10.1021/jo0015974 |
| 97. | Li, L.-S.; Wu, Y.-L. Tetrahedron Lett. 2002, 43, 2427–2430. doi:10.1016/S0040-4039(02)00290-3 |
| 98. | Ma, M.; Li, C.; Peng, L.; Xie, F.; Zhang, X.; Wang, J. Tetrahedron Lett. 2005, 46, 3927–3929. doi:10.1016/j.tetlet.2005.03.199 |
| 99. | Shimizu, H.; Murakami, M. Chem. Commun. 2007, 2855–2857. doi:10.1039/b704105e |
| 3. | Baxendale, I. R.; Pitts, M. R. Chim. Oggi 2006, 24 (3), 41–45. |
| 4. | Baxendale, I. R.; Ley, S. V. Heterogeneous Reactions. In New Avenues to Efficient Chemical Synthesis, Emerging Technologies; Seeberger, P. H.; Blume, T., Eds.; Springer-Verlag: Berlin, Heidelberg, 2007; pp 151–185. |
| 5. | Baxendale, I. R.; Hayward, J. J.; Ley, S. V. Comb. Chem. High Throughput Screening 2007, 10, 802–836. doi:10.2174/138620707783220374 |
| 6. |
Baxendale, I. R.; Hayward, J. J.; Lanners, S.; Ley, S. V.; Smith, C. D. Heterogeneous Reactions. In Microreactors in Organic Synthesis and Catalysis; Wirth, T., Ed.; Wiley-VCH: Weinheim, 2008; pp 84–122.
Chapter 4.2. |
| 7. | Jas, G.; Kirschning, A. Chem.–Eur. J. 2003, 9, 5708–5723. doi:10.1002/chem.200305212 |
| 8. | Hodge, P. Curr. Opin. Chem. Biol. 2003, 7, 362–373. doi:10.1016/S1367-5931(03)00052-8 |
| 9. |
Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem. 2004, 116, 410–451. doi:10.1002/ange.200300577
Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577 |
| 10. | Kirschning, A.; Solodenko, W.; Mennecke, K. Chem.–Eur. J. 2006, 12, 5972–5990. doi:10.1002/chem.200600236 |
| 11. | Ahmed-Omer, B.; Brandt, J. C.; Wirth, T. Org. Biomol. Chem. 2007, 5, 733–740. doi:10.1039/b615072a |
| 12. | Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c |
| 13. | Glasnov, V. T. N.; Kappe, C. O. Macromol. Rapid Commun. 2007, 28, 395–410. doi:10.1002/marc.200600665 |
| 14. | Benito-López, F.; Egberink, R. J. M.; Reinhoudt, D. N.; Verboom, W. Tetrahedron 2008, 64, 10023–10040. doi:10.1016/j.tet.2008.07.108 |
| 73. | Audrain, H.; Thorhauge, J.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2000, 65, 4487–4497. doi:10.1021/jo9918596 |
| 74. | Peng, Z.-H.; Woerpel, K. A. Org. Lett. 2002, 4, 2945–2948. doi:10.1021/ol026343e |
| 75. | Yu, S.; Saenz, J.; Srirangam, J. K. J. Org. Chem. 2002, 67, 1699–1702. doi:10.1021/jo016131f |
| 76. | Griesbeck, A. G.; Bondock, S.; Lex, J. Org. Biomol. Chem. 2004, 2, 1113–1115. doi:10.1039/b401990c |
| 77. | Sun, Y.; Wan, X.; Wang, J.; Meng, Q.; Zhang, H.; Jiang, L.; Zhang, Z. Org. Lett. 2005, 7, 5425–5427. doi:10.1021/ol052212c |
| 78. | Zhang, W.; Shi, M. Chem. Commun. 2006, 1218–1220. doi:10.1039/b516467b |
| 79. | Howard, B. E.; Woerpel, K. A. Org. Lett. 2007, 9, 4651–4653. doi:10.1021/ol702148x |
| 80. | Kratzer, R.; Nidetzky, B. Chem. Commun. 2007, 1047–1049. doi:10.1039/b616475g |
| 81. | Ntaganda, R.; Milovic, T.; Tiburcio, J.; Thadani, A. N. Chem. Commun. 2008, 4052–4054. doi:10.1039/b808302a |
| 56. | Cohen, B. J.; Kraus, M. A.; Patchornik, A. J. Am. Chem. Soc. 1977, 99, 4165–4167. doi:10.1021/ja00454a050 |
| 57. | Cohen, B. J.; Kraus, M. A.; Patchornik, A. J. Am. Chem. Soc. 1981, 103, 7620–7629. doi:10.1021/ja00415a034 |
| 58. | Patchornik, A. In Proc. IUPAC, I. U. P. A. C., Macromol. Symp., 28th, 1982, University of Massachusetts, Amherst, July 12–16, 1982; IUPAC: Oxford, 1982; p 85. |
| 59. | Brown, S. D.; Armstrong, R. W. J. Am. Chem. Soc. 1996, 118, 6331–6332. doi:10.1021/ja961203j |
| 60. | Hu, Y.; Baudart, S.; Porco, J. A., Jr. J. Org. Chem. 1999, 64, 1049–1051. doi:10.1021/jo981874v |
| 61. | Studer, A.; Hadida, S.; Ferritto, R.; Kim, S.-Y.; Jeger, P.; Wipf, P.; Curran, D. P. Science 1997, 275, 823–826. doi:10.1126/science.275.5301.823 |
| 62. | Flynn, D. L.; Crich, J. Z.; Devraj, R. V.; Hockerman, S. L.; Parlow, J. J.; South, M. S.; Woodard, S. J. Am. Chem. Soc. 1997, 119, 4874–4881. doi:10.1021/ja963462e |
| 63. |
Curran, D. P. Angew. Chem. 1998, 110, 1230–1255. doi:10.1002/(SICI)1521-3757(19980504)110:9<1230::AID-ANGE1230>3.0.CO;2-Y
Angew. Chem., Int. Ed. 1998, 37, 1174–1196. doi:10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P |
| 64. |
Bosanac, T.; Yang, J.; Wilcox, C. S. Angew. Chem. 2001, 113, 1927–1931. doi:10.1002/1521-3757(20010518)113:10<1927::AID-ANGE1927>3.0.CO;2-#
Angew. Chem., Int. Ed. 2001, 40, 1875–1879. doi:10.1002/1521-3773(20010518)40:10<1875::AID-ANIE1875>3.0.CO;2-5 |
| 65. |
Ley, S. V.; Massi, A.; Rodríguez, F.; Horwell, D. C.; Lewthwaite, R. A.; Pritchard, M. C.; Reid, A. M. Angew. Chem. 2001, 113, 1088–1090. doi:10.1002/1521-3757(20010316)113:6<1088::AID-ANGE10880>3.0.CO;2-#
Angew. Chem., Int. Ed. 2001, 40, 1053–1055. doi:10.1002/1521-3773(20010316)40:6<1053::AID-ANIE10530>3.0.CO;2-D |
| 66. | Galante, A.; Lhoste, P.; Sinou, D. Tetrahedron Lett. 2001, 42, 5425–5427. doi:10.1016/S0040-4039(01)01055-3 |
| 67. | Yoshida, J.-i.; Itami, K. Chem. Rev. 2002, 102, 3693–3716. doi:10.1021/cr0103524 |
| 68. | Dobbs, A. P.; McGregor-Johnson, C. Tetrahedron Lett. 2002, 43, 2807–2810. doi:10.1016/S0040-4039(02)00322-2 |
| 69. | Lan, P.; Porco, J. A., Jr.; South, M. S.; Parlow, J. J. J. Comb. Chem. 2003, 5, 660–669. doi:10.1021/cc030028h |
| 70. | Siu, J.; Baxendale, I. R.; Lewthwaite, R. A.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3140–3160. doi:10.1039/b503778f |
| 71. | Curran, D. P.; Wang, X.; Zhang, Q. J. Org. Chem. 2005, 70, 3716–3719. doi:10.1021/jo050116j |
| 72. | Mothana, S.; Chahal, N.; Vanneste, S.; Hall, D. G. J. Comb. Chem. 2007, 9, 193–196. doi:10.1021/cc060149s |
| 90. | Nimitz, J. S.; Mosher, H. S. J. Org. Chem. 1981, 46, 211–213. doi:10.1021/jo00314a057 |
| 91. | Creary, X.; Mehrsheikh-Mohammadi, M. E. J. Org. Chem. 1986, 51, 2664–2668. doi:10.1021/jo00364a009 |
| 92. | Babudri, F.; Fiandanese, V.; Marchese, G.; Punzi, A. Tetrahedron 1996, 52, 13513–13520. doi:10.1016/0040-4020(96)00805-8 |
| 38. | Baxendale, I. R.; Deeley, J.; Griffiths-Jones, C. M.; Ley, S. V.; Saaby, S.; Tranmer, G. K. Chem. Commun. 2006, 2566–2568. doi:10.1039/b600382f |
| 39. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Org. Lett. 2006, 8, 5231–5234. doi:10.1021/ol061975c |
| 40. | Smith, C. J.; Iglesias-Sigüenza, F. J.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 2758–2761. doi:10.1039/b709043a |
| 41. | Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k |
| 42. | Hornung, C. H.; Mackley, M. R.; Baxendale, I. R.; Ley, S. V. Org. Process Res. Dev. 2007, 11, 399–405. doi:10.1021/op700015f |
| 43. | Baumann, M.; Baxendale, I. R.; Ley, S. V. Synlett 2008, 2111–2114. doi:10.1055/s-2008-1078026 |
| 44. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N.; Smith, C. D. Org. Biomol. Chem. 2008, 6, 1587–1593. doi:10.1039/b801634h |
| 45. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N.; Smith, C. D.; Tierney, J. P. Org. Biomol. Chem. 2008, 6, 1577–1586. doi:10.1039/b801631n |
| 46. | Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tamborini, L.; Voica, A.-F. J. Comb. Chem. 2008, 10, 851–857. doi:10.1021/cc800070a |
| 47. | Jas, G.; Kirschning, A. Chem.–Eur. J. 2003, 9, 5708–5723. doi:10.1002/chem.200305212 |
| 48. | Bernstein, D.; France, S.; Wolfer, J.; Lectka, T. Tetrahedron: Asymmetry 2005, 16, 3481–3483. doi:10.1016/j.tetasy.2005.09.014 |
| 49. | Bonfils, F.; Cazaux, I.; Hodge, P.; Caze, C. Org. Biomol. Chem. 2006, 4, 493–497. doi:10.1039/b515241k |
| 50. | Wiles, C.; Watts, P.; Haswell, S. J. Tetrahedron Lett. 2006, 47, 5261–5264. doi:10.1016/j.tetlet.2006.05.157 |
| 51. | Dräger, G.; Kiss, C.; Kunz, U.; Kirschning, A. Org. Biomol. Chem. 2007, 5, 3657–3664. doi:10.1039/b712804e |
| 52. | Burguete, M. I.; Cornejo, A.; García-Verdugo, E.; Gil, M. J.; Luis, S. V.; Mayoral, J. A.; Martínez-Merino, V.; Sokolova, M. J. Org. Chem. 2007, 72, 4344–4350. doi:10.1021/jo070119r |
| 53. | Solodenko, W.; Jas, G.; Kunz, U.; Kirschning, A. Synthesis 2007, 583–589. doi:10.1055/s-2007-965877 |
| 54. | Odedra, A.; Geyer, K.; Gustafsson, T.; Gilmour, R.; Seeberger, P. H. Chem. Commun. 2008, 3025–3027. doi:10.1039/b803715a |
| 82. | Peet, N. P.; Burkhart, J. P.; Angelastro, M. R.; Giroux, E. L.; Mehdi, S.; Bey, P.; Kolb, M.; Neises, B.; Schirlin, D. J. Med. Chem. 1990, 33, 394–407. doi:10.1021/jm00163a063 |
| 83. | Patel, D. V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C. A.; Smith, S. A.; Petrillo, E. W., Jr. J. Med. Chem. 1993, 36, 2431–2447. doi:10.1021/jm00069a001 |
| 84. | Li, Z.; Patil, G. S.; Golubski, Z. E.; Hori, H.; Tehrani, K.; Foreman, J. E.; Eveleth, D. D.; Bartus, R. T.; Powers, J. C. J. Med. Chem. 1993, 36, 3472–3480. doi:10.1021/jm00074a031 |
| 85. | Koutek, B.; Prestwich, G. D.; Howlett, A. C.; Chin, S. A.; Salehani, D.; Akhavan, N.; Deutsch, D. G. J. Biol. Chem. 1994, 269, 22937–22940. |
| 86. | Conde-Frieboes, K.; Reynolds, L. J.; Lio, Y.-C.; Hale, M. R.; Wasserman, H. H.; Dennis, E. A. J. Am. Chem. Soc. 1996, 118, 5519–5525. doi:10.1021/ja953553w |
| 87. | Otto, H.-H.; Schirmeister, T. Chem. Rev. 1997, 97, 133–172. doi:10.1021/cr950025u |
| 88. | Choe, Y.; Brinen, L. S.; Price, M. S.; Engel, J. C.; Lange, M.; Grisostomi, C.; Weston, S. G.; Pallai, P. V.; Cheng, H.; Hardy, L. W.; Hartsough, D. S.; McMakin, M.; Tilton, R. F.; Baldino, C. M.; Craik, C. S. Bioorg. Med. Chem. 2005, 13, 2141–2156. doi:10.1016/j.bmc.2004.12.053 |
© 2009 Palmieri et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)