Abstract
Methodology has been developed for laying down a thin gold-on-silver film on the inner surface of glass capillaries for the purpose of catalysing benzannulation reactions. The cycloaddition precursors are flowed through these capillaries while the metal film is being heated to high temperatures using microwave irradiation. The transformation can be optimized rapidly, tolerates a wide number of functional groups, is highly regioselective, and proceeds in good to excellent conversion.

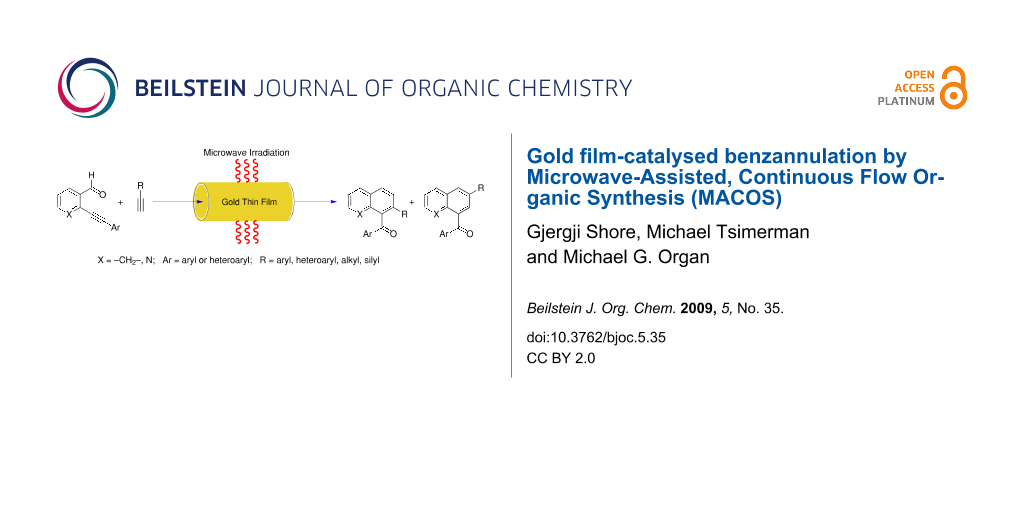
Graphical Abstract
Introduction
Microwave-assisted organic synthesis (MAOS) has had a significant impact on organic and medicinal chemistry by dramatically shortening reaction times, producing cleaner product mixtures, and making high-energy transformations routine that might otherwise be avoided [1,2]. Within the last five years, microwave technology has also been applied to reactions performed in a flowed format [3,4]. Flowed chemical synthesis offers numerous advantages over traditional batch-reactor technology [5-24]. Independent inlet streams allow reactive intermediates to be kept separate until brought together in miniscule amounts to react immediately; this rapidly depletes the starting materials and continuously physically moves the product away from the infusing stream. In batch reactors, product molecules form in the presence of a vast excess of starting materials that can lead to significant byproduct formation. Further, a moving synthesis platform allows for in-line analysis and instantaneous changes to reaction conditions for process optimization that can be automated readily. To gain the full advantage of working in flow, reactions should proceed very rapidly and ideally reach completion during the time in which the reactants reside in the flow tube. Microwave heating has been used to drive a wide variety of reactions to high levels of completion in a flowed format [5-24].
We have demonstrated that thin-metal films on the walls of narrow tubes can impart tremendous rate accelerations on flowed reactions that are being simultaneously microwaved [25-28]. These accelerations can be a result of direct catalysis by the film itself, the tremendous temperatures attainable by microwaved metal films, or a combination of the two. In cross coupling processes, the film has been demonstrated itself to be capable of catalyzing Suzuki–Miyaura and Heck reactions, i.e., Pd-thin films promote these transformations without any additional catalyst being added to the reactant stream(s) that enter the flow tube [28]. Without microwave irradiation, the above-described cross-coupling reactions did not proceed indicating that there is not only a catalytic effect, but also a pivotal heating effect supplied by the film. More recently we have shown that a gold film in the same flow reactor is highly effective for hydrosilylation reactions [25] and we were interested both to expand the use of gold films in synthesis using MACOS and in exploring complex, multi-step catalytic processes in flow.
The benzannulation reaction between aromatic carbonyls and alkynes has received increasing attention since 2002 [29,30]. This transformation has been shown to be promoted by Lewis acids, copper complexes [31] and various gold species, including Au(I)X, Au(III)X3, [32-34] and Au nanoparticles dispersed on different supports. For Au(III) catalysts, the reaction has been proposed to proceed via the formation of no less than four organogold intermediates and/or complexes (Figure 1).
![[1860-5397-5-35-1]](/bjoc/content/figures/1860-5397-5-35-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Mechanism of Au(III)-catalyzed benzannulation between aromatic carbonyls and alkynes.
Figure 1: Mechanism of Au(III)-catalyzed benzannulation between aromatic carbonyls and alkynes.
Further, it has been shown computationally by Straub that Au(I) and Au(III) can perform both this and related catalytic cycles with similar energy profiles [35]. This blurs the distinction of the two pathways and raises the possibility that the actual active species in these transformations could be either species, providing that the possibility exists for the starting Au complex to be oxidized or reduced under a specific set of reaction conditions. Given the complexity of this process, we thought that benzannulation would be an ideal transformation to investigate the use of gold films for MACOS.
Results and Discussion
Our investigation started with an assessment of different types of metal films for their utility in benzannulation. The morphology of Cu, Pd, Ag, and Au films can be compared in the scanning electron microscopy (SEM) images in Figure 2. The films are generally prepared in a reducing environment (see Experimental), thus they are expected to be largely M(0), which has been confirmed by energy-dispersive X-ray (EDX) analysis of the films (e.g., see panel f in Figure 2).
![[1860-5397-5-35-2]](/bjoc/content/figures/1860-5397-5-35-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray analysis of the metal films used in this benzannulation study. Panels a–e are scanning-electron micrographs (SEM) of all films taken at 30,000 × magnification: a) Cu, b) Pd, c) Ag, d) Au layered on a base film of Ag. Panel e shows the side view of the Au film attached to the glass capillary wall (bottom of image), between which is sandwiched a barely-visible thin Ag coating. Panel f is the energy-dispersive X-ray (EDX) spectrum of the Au film from panel d; note the small level of Ag that is present that helps to anchor the Au to the surface of the glass.
Figure 2: X-ray analysis of the metal films used in this benzannulation study. Panels a–e are scanning-electr...
However, film preparation was not conducted under a strictly inert environment, thus oxygen and/or water in the air could play a role in the film’s composition leading to the formation metal oxides, for example, on the surface. Palladium films had the greatest porosity (panel b) while silver films (panel c) were the most dense and uniform. Gold films showed poor adhesion to the glass and poor performance when subjected to the rigors of MACOS. However, these properties were dramatically improved when the same gold film was laid down on a transparently-thin silver layer that coated the capillary wall. With these metal-film-coated capillaries in hand, benzannulation was investigated using the substrates shown in Table 1.
Table 1: Optimization Studies for Benzannulation Reaction using MACOS.
![[Graphic 1]](/bjoc/content/inline/1860-5397-5-35-i1.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Film | Heat Source | Temp. (°C) | Percent Conversiona | Percent Yieldb | 3a : 4a |
|---|---|---|---|---|---|---|
| 1 | Cu | microwave | 240 | 5 | 4 | ND |
| 2 | Pd | microwave | 240 | 0 | 0 | – |
| 3 | Ag | microwave | 240 | 0 | 0 | – |
| 4 | Au on Au | microwave | 220 | 75 | ND | |
| 5 | Au on Ag | microwave | 240 | 90 | 78 | 3 : 1 |
| 6 | Au on Ag | microwave | 190 | 68 | ND | ND |
| 7 | Au on Ag | oil bathc | 190 | 14 | ND | ND |
aPercent conversion was determined by 1H NMR spectroscopy by comparing the ratio of product peaks (3 and/or 4) to the starting aldehyde (1) from aliquots taken directly from the eluent stream. bPercent yield was determined by collecting a specific volume of eluent that contained a known amount of starting materials, and purifying the material by silica gel flash chromatography. cThe metal-film-coated capillary was left in the bath for 20 min to ensure that the film was at temperature prior to flowing the reaction mixture through it.
This reaction is reported most widely using homogeneous and heterogeneous gold catalysts, and indeed we found that a pure Au film provided good conversion for a short period (Table 1, entry 4), although the film had a short lifespan. Films of palladium (entry 2) and silver (entry 3) showed no conversion of starting materials at all. Surprisingly, copper, which is reported to be a suitable catalyst for benzannulation, could not be optimized beyond the formation of only trace amounts of product (entry 1). We experimented with the use of bimetallic films and found that optimal film performance was achieved by laying down a porous gold film on top of a thin silver mirror. When run at a high temperature (240 °C, entry 5), as read by the IR sensor in the Biotage Initiator microwave, excellent conversion was achieved that diminished as the temperature was reduced (e.g., 190 °C, entry 6). It is noteworthy that under batch conditions, these reactions are reported to require up to 6 h to obtain similar conversion levels [7] to those obtained in less than 60 s using MACOS, which is the residence time of any reactant plug in the flow reactor.
To probe any special effect brought about by the microwave in this transformation, the identical transformation in entry 6 was performed using an oil bath to achieve the same temperature (i.e., 190 °C, entry 7) and the conversion was dramatically reduced from 68 to 14 percent. It is possible that superior microwave conversion is the result of localized ‘hot spots’ in the film that are potentially well above the bulk temperature recorded by the IR sensor when microwave irradiation is used. It has been proposed that steps or angles in the surface of the metal film can serve as an antenna resulting in higher levels of microwave absorption in those areas causing higher than normal temperatures. In an oil bath (or oven), only the bulk temperature of the external heating source can be achieved in the film. If this is the case, and localized areas of potentially extreme temperature are required for optimal reaction performance, this would be preferable to heating the entire film to these higher temperatures, which would lead to faster film and product decomposition.
With optimized conditions in hand, we set out to examine the substrate scope of this reaction using capillaries lined with the gold-on-silver films (Table 2).
Table 2: Benzannulation reactions with Au on Ag-coated capillaries using MACOS.
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-35-i2.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R– | –X– | R1– | Percent Conversiona | Percent Yieldb | 3 : 4 |
|---|---|---|---|---|---|---|
| a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-5-35-i3.svg?max-width=637&scale=1.0)
|
–CH– |
![[Graphic 4]](/bjoc/content/inline/1860-5397-5-35-i4.svg?max-width=637&scale=1.0)
|
90 | 78 | 75 : 25 |
| b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-5-35-i5.svg?max-width=637&scale=1.0)
|
–CH– | (CH3)3Si– | 75 | 62 | 4 only |
| c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-5-35-i6.svg?max-width=637&scale=1.0)
|
–CH– |
![[Graphic 7]](/bjoc/content/inline/1860-5397-5-35-i7.svg?max-width=637&scale=1.0)
|
62 | 52 | 58 : 42 |
| d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-5-35-i8.svg?max-width=637&scale=1.0)
|
–CH– |
![[Graphic 9]](/bjoc/content/inline/1860-5397-5-35-i9.svg?max-width=637&scale=1.0)
|
76 | 62 | 3 only |
| e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-5-35-i10.svg?max-width=637&scale=1.0)
|
–CH– |
![[Graphic 11]](/bjoc/content/inline/1860-5397-5-35-i11.svg?max-width=637&scale=1.0)
|
72 | 60 | 4 only |
| f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-5-35-i12.svg?max-width=637&scale=1.0)
|
–N– |
![[Graphic 13]](/bjoc/content/inline/1860-5397-5-35-i13.svg?max-width=637&scale=1.0)
|
78 | 64 | 70 : 30 |
| g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-5-35-i14.svg?max-width=637&scale=1.0)
|
–N– |
![[Graphic 15]](/bjoc/content/inline/1860-5397-5-35-i15.svg?max-width=637&scale=1.0)
|
68 | 54 | 4 only |
| h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-5-35-i16.svg?max-width=637&scale=1.0)
|
–N– | (CH3)3Si– | 65 | 58 | 4 only |
| i |
![[Graphic 17]](/bjoc/content/inline/1860-5397-5-35-i17.svg?max-width=637&scale=1.0)
|
–N– |
![[Graphic 18]](/bjoc/content/inline/1860-5397-5-35-i18.svg?max-width=637&scale=1.0)
|
65 | 52 | 4 only |
| j |
![[Graphic 19]](/bjoc/content/inline/1860-5397-5-35-i19.svg?max-width=637&scale=1.0)
|
–N– |
![[Graphic 20]](/bjoc/content/inline/1860-5397-5-35-i20.svg?max-width=637&scale=1.0)
|
50 | 40 | 4 only |
aPercent conversion was determined by 1H NMR spectroscopy by comparing the ratio of product peaks (3 and/or 4) to the starting aldehyde (1) from aliquots taken directly from the eluent stream. bPercent yield was determined by collecting a specific volume of eluent that contains a known amount of starting materials, and purifying the material by silica gel flash chromatography.
The reaction was applicable to the hydrocarbon starting materials tried (e.g., entries a, b, c) and was also useful for the preparation of heterocycle-containing molecules such as pyridines and thiophenes (e.g., entries e–j). A survey of different functional groups revealed a broad tolerance including silyl groups (entries b and h), halides (entries g and i), ethers, (entry c), amides (entry j), and even free carboxylic acids (entry e). In all cases where reactions did not fully complete, benzaldehyde (1) accounted for the mass balance; crude 1H NMR spectra were very clean showing only residual 1, product and trace by products. In most cases, the reactions displayed a high level of regioselectivity.
All of the above runs were conducted on approximately 700 μL of an infusing starting material solution (35 min per run), which, after purification, generated ~70–80 mg of final product. Such quantities are ample for evaluation in biological screens. However, larger quantities of a compound can be required, so using the substrates in Table 2, entry a, a larger-scale benzannulation was performed. To improve efficiency and to reduce solvent consumption (and hence waste production) the concentration of both starting materials was tripled. After running the reactor for 90 min, three quarters of a gram of product (3a and 4a, 3:1) were collected. Under these conditions the concentration of alkyne 2a was 4.5 M, demonstrating that MACOS can easily handle concentrations that are far above the typical levels (0.1–0.5 M) commonly used in conventionally-heated batch reactors.
The lifetime and durability of the film under the optimized reaction conditions was examined. The transformation in Table 2, entry a was followed over the course of 2 h; reaction performance is optimal at the beginning and slowly, but steadily erodes. Visibly, the film darkens over time, which we attribute to small amounts of starting materials and/or products that char over time on the surface of the hot film; this may lead to the blockage of reaction sites. Additionally, the thin-walled capillary glass softened and in some cases failed after 90 min of exposure to the hot film. While bulk manufacturing of very large quantities is not yet attainable with the current reactor design, the device is very suitable for preparing library collections of 50–750 mg of product for evaluation in medicinal chemistry or materials science applications.
Conclusion
Glass capillaries internally-lined with thin-metal films of gold, copper, silver, and gold-on-silver were prepared and evaluated for their catalytic activity in the microwave-assisted, continuous flow benzannulation of aryl, alkyl and silylalkynes with alkynylbenzaldehydes. Only the gold-on-silver films possessed both the high level of catalytic activity and suitable physical robustness to prepare quantities of product suitable, for example, for biological or material science evaluation purposes (up to ~700 mg). The reaction showed wide functional group tolerance and good to excellent regioselectivity in the cycloaddition. We are now evaluating new materials to replace the glass tube with the plan of making the process more sustainable.
Experimental
Microwave irradiation experiments
All MACOS experiments were performed in 1700 μm (ID) borosilicate capillaries, using a single mode Biotage Smith Creator Synthesizer, operating at a frequency of 2.45 GHz with irradiation power from 0 to 300 W. The capillary was fed reactants from Hamilton gastight syringes attached to a Harvard 22 syringe pump pre-set to the desired flow rate. The system was connected to a sealed collection vial, where a pressurized air line (75 psi) was attached to create backpressure. The temperatures reported were measured off the surface of the capillaries by the IR sensor built into the microwave chamber. All reagents and solvents were purchased from commercial sources and used without additional purification. Column chromatography purifications were carried out using the flash technique on silica gel 60 (200–400 mesh). 1H NMR spectroscopy was run using a Bruker Advance 400 MHz instrument and all spectra were calibrated to the signal from the residual proton of the deuterated chloroform solvent (7.26 ppm); 13C NMR spectra were calibrated to the middle carbon signal of the triplet for deuterated chloroform (77.00 ppm).
General procedure for the benzannulation reactions by MACOS
A stock solution containing the acetylenic aldehyde (0.5 mmol, 1.0 equiv.) and alkyne (1.5 mmol, 3.0 equiv) in 0.7–0.8 mL 1,2-dichloro benzene (total mixture volume is 1.0 mL) was prepared and loaded into a Hamilton gastight syringe. The tubing was primed with 1,2-dichloro benzene and the syringe was connected to the reactor system with the aid of Microtight fittings after which it was placed in a Harvard 22 syringe pump that was set to deliver 20 μL/min. The single mode microwave was programmed to heat constantly; the power level was controled manually so as to keep the temperature constant at the specified levels. The effluent from the reactor was fed into a sealed vial and analyzed directly by 1H NMR spectroscopy immediately after the reaction. Typically, 0.7–0.8 mL of the crude reaction mixture was collected and the product was purified by silica gel column chromatography.
General procedure for creating the gold-on-silver film coating inside of 1700 micron (ID) capillaries
Tollens’ reagent (0.5 mL) was mixed with 0.5 mL of a 5% D-glucose solution into a 2 mL vial. The 1700 μm capillaries (ID) were filled with this mixture, capped at both ends and left to develop at rt. After the Ag coating was fully developed (15 min), the capillaries were rinsed with acetone and placed inside a muffle furnace for calcination at 500 °C (3 × 1 min).
The gold-coating solution was prepared by mixing a 0.4 mmol/mL aqueous solution of AuCl3 (0.5 mL) with a 2% aqueous solution of sodium citrate (0.5 mL). The Ag-lined capillaries were filled with the mixture, capped at both ends, and left to develop at rt for an additional 30 min. After emptying the capillaries and rinsing them with acetone, they were calcinated at 500°C (3 × 1min) before use in MACOS.
Tollen’s reagent was prepared as follows: 2.0 mL of 4M NaOH was added drop-wise to 20 mL of a 3% AgNO3 solution, forming a gray precipitate that was titrated with a 4M solution of NH4OH until the solution became clear.
General procedure for synthesis of benzaldehydes [36]
![[Graphic 21]](/bjoc/content/inline/1860-5397-5-35-i21.svg?max-width=637&scale=1.18182)
A 10 mL round-bottom flask was charged with Pd(PPh3)4 (0.0375 mmol), CuI (0.075 mmol), and purged with argon. To it were added consecutively: THF (6 mL), Et3N (1.5 mmol), the aryl bromide (0.75 mmol), and the alkyne (0.9 mmol). The solution was then heated to 70 °C and stirred for 6–13 h (until the reaction was judged complete by tlc analysis). At the end of the reaction, the mixture was diluted with H2O (10 mL) and extracted with ethyl acetate (3×). The combined organic layers were dried over anhydrous Na2SO4, concentrated, the residue dry loaded onto silica gel and purified by column chromatography.
![[Graphic 22]](/bjoc/content/inline/1860-5397-5-35-i22.svg?max-width=637&scale=1.18182)
2-(2-Phenylethynyl)benzaldehyde (1a). Following the general procedure for the synthesis of benzaldehydes, column chromatography (10% diethyl ether in pentane) provided 141 mg of product as a pale-brown oil (91%, yield). 1H NMR (300 MHz, CDCl3): δ 10.67 (s, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 4.5 Hz, 1H), 7.57 (m, 3H), 7.44 (t, J = 7.2 Hz, 1H), 7.41–7.37 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 191.6, 135.8, 133.8, 133.2, 131.7, 129.1, 128.6, 128.5, 127.3, 126.9, 122.4, 96.4, 85.0. Spectra matched that found in the literature [37].
![[Graphic 23]](/bjoc/content/inline/1860-5397-5-35-i23.svg?max-width=637&scale=1.18182)
2-[2-(Thiophen-3-yl)ethynyl]benzaldehyde (1d). Following the general procedure for the synthesis of benzaldehydes, column chromatography (10% diethyl ether in pentane) provided 142 mg of the product as a pale-yellow oil (89%, yield). 1H NMR (300 MHz, CDCl3): δ 10.63 (s, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.66–7.51 (m, 3H), 7.43 (t, J = 7.8 Hz, 1H), 7.34 (dd, J = 5.1, 3.0 Hz, 1H), 7.23 (dd, J = 5.1, 1.2 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ 191.7, 135.8, 133.8, 133.1, 129.7 (two carbons overlap), 128.5, 127.3, 126.9, 125.8, 121.4, 91.5, 84.6. HRMS Calcd. for C13H8OS: 212.0296; found: 212.0302: Anal. Calcd. for C13H8OS: C, 73.56; H, 3.80; found C, 73.82; H, 3.57.
![[Graphic 24]](/bjoc/content/inline/1860-5397-5-35-i24.svg?max-width=637&scale=1.18182)
2-(2-Phenylethynyl)nicotinaldehyde (1f). Following the general procedure for the synthesis of benzaldehydes, column chromatography (30% diethyl ether in pentane) provided 145 mg of product as a dark-yellow solid (93%, yield). Mp = 91–92 °C. 1H NMR (400 MHz, CDCl3): δ 10.70 (s, 1H), 8.85 (d, J = 3.6 Hz, 1H), 8.24 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 6.8 Hz, 2H), 7.55–7.30 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 190.8, 154.5, 146.1, 134.9, 132.2, 131.8, 129.9, 128.6, 123.2, 121.2, 96.1, 84.6. HRMS Calcd. for C14H9NO 207.0684; found 207.0685. Anal. Calcd. for C14H9NO: C, 81.14; H, 4.38; found C, 81.43; H, 4.60. Spectra matched that found in the literature [38].
![[Graphic 25]](/bjoc/content/inline/1860-5397-5-35-i25.svg?max-width=637&scale=1.18182)
3-Ethynyl-N,N-diisopropylbenzamide (2j). A 25 mL round-bottom flask was purged with argon, cooled to 0 °C, and 2.4 mL of dry THF and 157 μL (1.8 mmol) of oxalyl chloride were added. After 20 min., a cooled (0 °C) solution of 3-ethynyl-benzoic acid (1.71 mmol, 250 mg) in 6 mL of dry THF was added over 10 min. When the addition of acid was complete, 3 drops of DMF were added to the mixture and bubbling was observed. After stirring for 30 min. at 0 °C, the solution was stirred an additional 30 min. at rt, and then cooled back to 0 °C. Into a separate 25 mL round-bottom flask at 0 °C were added: (iPr)2NH (1.8 mmol, 260 μL), Et3N (1.8 mmol, 250 μL), and 2 mL of dry THF. The contents of the original flask were then added to this flask where upon fuming was observed and a yellow precipitate began to form. The reaction was warmed to rt, stirred for 16 h, and quenched by the addition of 10 mL of water. After stirring for 10 min., the contents were then transferred to a separatory funnel and ethyl acetate was added. The separated organic layer was washed with sat NaHCO3 and the aqueous layer was back-extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and filtered. Following solvent removal in vacuo, the yellow solid residue was dry-loaded onto silica gel and purified by column chromatography using a gradient elution (5% ethyl acetate in pentane to 15% ethyl acetate in pentane, Rf = 0.5) to give 390 mg product as a white solid (99% yield). Mp = 107–108 °C. 1H NMR (400 MHz, CDCl3): δ 7.49 (d, J = 7.6 Hz, 1H), 7.43 (s, 1H), 7.36 (t, J = 8.4 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 7.32 (d, 2H), 3,09 (s, 1H), 1.33 (d, 12H). 13C NMR (150 MHz, CDCl3, 10 °C): δ 169.8, 139.0, 132.2, 129.1, 128.6, 125.9, 122.4, 82.9, 77.9, 51.0, 45.9, 20.7, 20.6. HRMS Calcd. for C15H19NO: 229.1467; found: 229.1466. Anal. Calcd. for C15H19NO: C, 78.56; H, 8.35; found C, 78.61; H, 8.71.
![[Graphic 26]](/bjoc/content/inline/1860-5397-5-35-i26.svg?max-width=637&scale=1.18182)
Phenyl-(2-phenylnaphthalen-1-yl)-methanone (3a). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-benzaldehyde (1a) and phenylacetylene were reacted and 700 μL of the crude reaction mixture were collected. Purification by flash chromatography (15% ethyl acetate in hexane) afforded 62 mg of 3a and 20 mg of 4a as yellow oils (78% combined yield, minor isomer reported below). 1H NMR (400 MHz, CDCl3): δ 8.2 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.65 (m, 2H), 7.58 (d, J = 8.3 Hz, 1H), 7.35–7.52 (m, 5H), 7.15–7.30 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 199.8, 140.4, 137.9, 137.6, 137.2, 133.4, 132.5, 130.5, 129.5, 129.4, 129.3, 128.3, 128.1, 128.0, 127.6, 127.2, 127.0, 126.4, 125.5. Spectra matched that found in the literature [39].
![[Graphic 27]](/bjoc/content/inline/1860-5397-5-35-i27.svg?max-width=637&scale=1.18182)
Phenyl-(3-phenylnaphthalen-1-yl)-methanone (4a). 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 1.1 Hz, 1H), 7.98 (d, J = 8.2 Hz, 1H), 7. 90 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 1.1 Hz, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.50 (tt, J = 7.4, 1.0 Hz, 1H), 7.37–7.47 (m, 6H), 7.30 (tt, J = 7.4, 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 197.7, 139.9, 138.2, 137.2, 137.0, 134.2, 133.4, 130.5, 130.1, 129.0, 128.8, 128.6, 128.3, 127.8, 127.2, 127.0, 126.9, 126.7, 125.7. Spectra matched that found in the literature [40].
![[Graphic 28]](/bjoc/content/inline/1860-5397-5-35-i28.svg?max-width=637&scale=1.18182)
Phenyl-(3-trimethylsilanyl-naphthalen-1-yl)-methanone (4b). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-benzaldehyde (1a) and TMS-acetylene were reacted and 740 μL of the crude reaction mixture were collected. Purification by flash chromatography (15% ethyl acetate in hexane) afforded 68.9 mg of 4b as a white solid (62% yield). Mp = 86–87 °C (lit. [34]: 88 °C). 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 1.1 Hz, 1H), 8.05 (d, J = 8.2 Hz, 1H), 7.85–7.95 (m, 3H), 7.68 (d, J = 1.1Hz, 1H), 7.45–7.60 (m, 5H), 0.35 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 198.5, 138.3, 136.8, 136.5, 135.5, 133.3, 133.0, 131.6, 131.1, 130.5, 128.5, 128.3, 127.4, 126.4, 125.6, −1.20. Spectra matched that found in the literature [34].
![[Graphic 29]](/bjoc/content/inline/1860-5397-5-35-i29.svg?max-width=637&scale=1.18182)
[2-(Methoxymethyl)naphthalen-1-yl]-phenylmethanone (3c). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-benzaldehyde (1a) and methyl propargyl ether were reacted and 815 μL of the crude reaction mixture were collected. Purification by flash chromatography (15% ethyl acetate in pentane) afforded 58.0 mg of 3c and 4c isomers as colourless oils (52% combined yield, 58:42, respectively).
(3c) 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.84 (d, J = 8.1Hz, 2H), 7.53–7.66 (m, 3H), 7.37–7.52 (m, 4H), 4.50 (s, 2H), 3.23 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 199.2, 137.8, 135.7, 133.7, 133.3, 132.8, 130.4, 129.6, 129.4, 128.7, 128.2, 126.8, 126.2, 125.5, 125.4, 72.1, 58.3. Anal. Calcd. for C19H16O2: C, 82.58; H, 5.84; found, C, 82.34, H, 5.60. HRMS calcd. for C19H16O2: 276.1150; found 276.1154.
![[Graphic 30]](/bjoc/content/inline/1860-5397-5-35-i30.svg?max-width=637&scale=1.18182)
[3-(Methoxymethyl)naphthalen-1-yl]-phenylmethanone (4c). 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.1Hz, 1H), 7.98 (d, J = 1.1 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 8.1 Hz, 2H), 7.10 (t, J = 7.1 Hz, 1H), 7.58 (d, J = 1.1 Hz, 1H), 7.45–7.56 (m, 4H), 4.66 (s, 2H), 3.46 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 197.9, 138.2, 136.7, 134.4, 133.7, 133.3, 130.5, 130.4, 129.4, 128.5, 128.3, 127.3, 127.1, 126.7, 125.6, 74.2, 58.3. Anal. Calcd. for C19H16O2: C, 82.58; H, 5.84; found C, 82.39; H, 5.62. HRMS calcd. for C19H16O2: 276.1150; found 276.1153.
![[Graphic 31]](/bjoc/content/inline/1860-5397-5-35-i31.svg?max-width=637&scale=1.18182)
[2-(Phenyl)naphthalen-1-yl](thiophen-3-yl)-methanone (3d). Following the general MACOS benzannulation protocol, 2-(2-(thiophen-3-yl)-ethynyl)-benzaldehyde (1d) and phenylacetylene were reacted and 650 μL of the crude reaction mixture were collected. Purification by flash chromatography (15% ethyl acetate in hexane) afforded 63.7 mg of 3d as a pale-yellow oil (62% yield). 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J = 8.1 Hz, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.61 (d, J = 8.1 Hz, 1H), 7.48–7.58 (m, 3H), 7.38–7.47 (m, 3H), 7.21–7.34 (m, 3H), 7.12–7.15 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 198.6, 143.5, 140.3, 137.1, 136.4, 135.3, 132.4, 130.4, 129.6, 129.4, 128.3, 128.1, 127.6, 127.5, 127.3, 127.2, 126.4, 126.2, 125.5. Anal. Calcd. for C21H14OS: C, 80.22; H, 4.49; found C, 79.79; H, 4.13. HRMS calcd. for C21H14OS: 314.0765; found 314.0759.
![[Graphic 32]](/bjoc/content/inline/1860-5397-5-35-i32.svg?max-width=637&scale=1.18182)
3-[4-(Thiophene-3-carbonyl)naphthalen-2-yl]-benzoic acid (4e). Following the general MACOS benzannulation protocol, 2-(2-(thiophen-3-yl)-ethynyl)-benzaldehyde (1d) and 3-ethynylbenzoic acid were reacted and 700 μL of the crude reaction mixture were collected. Purification by flash chromatography (10% methanol in dichloromethane) afforded 75.0 mg of 4e as a yellow oil (60% yield). 1H NMR (400 MHz, CDCl3): δ 8.17 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.93–8.00 (m, 2H), 7.82 (d, J = 8.1 Hz, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.48–7.63 (m, 4H), 7.33–7.43 (m, 2H), 7.15 (s, 1H). 13C NMR (150 MHz, CDCl3): δ 192.7, 171.3, 143.4, 140.6, 136.7, 135.7, 135.3, 134.6, 132.6, 130.8, 130.3, 129.7, 129.4, 129.1, 128.4, 128.1, 127.3, 127.2, 127.0, 126.6, 126.4, 125.5. HRMS calcd. for C22H14O3S: 358.0664; found 358.0666.
![[Graphic 33]](/bjoc/content/inline/1860-5397-5-35-i33.svg?max-width=637&scale=1.18182)
Phenyl-(7-phenylquinolin-8-yl)-methanone (3f). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-nicotinaldehyde (1f) and phenyl acetylene were reacted and 780 μL of the crude reaction mixture were collected. Purification by flash chromatography (20% ethyl acetate in pentane) afforded 77.8 mg of 3f and 4f isomers as pale-yellow oils (64% combined yield). (3f) 1H NMR (400 MHz, CDCl3): δ 8.88 (dd, J = 4.1, 1.5 Hz, 1H), 8.25 (d, J = 8.1 Hz, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.66–7.73 (m, 3H), 7.40–7.47 (m, 4H), 7.20–7.33 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 198.8, 151.1, 146.6, 140.5, 139.3, 137.9, 137.4, 135.7, 133.0, 129.6, 129.3, 128.7, 128.6, 128.3, 128.2, 127.7, 127.0, 121.4. HRMS calcd. for C22H15NO: 309.1154; found 309.1145.
![[Graphic 34]](/bjoc/content/inline/1860-5397-5-35-i34.svg?max-width=637&scale=1.18182)
Phenyl-(6-phenylquinolin-8-yl)-methanone (4f). 1H NMR (400 MHz, CDCl3): δ 8.86 (dd, J = 4.1, 1.5 Hz, 1H), 8.29 (dd, J = 8.1, 1.5 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 8.03 (d, J = 2.0 Hz, 1H), 7.92 (dd, J = 8.1, 1.5 Hz, 2H), 7.75 (dd, J = 8.1, 1.5 Hz, 2H), 7.59 (t, J = 7.1 Hz, 1H), 7.49–7.56 (m, 2H), 7.40–7.49 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 197.7, 150.8, 145.6, 139.8, 139.5, 138.7, 137.7, 136.2, 133.3, 130.3, 129.1, 128.5, 128.3, 128.1, 127.9, 127.5, 127.1, 122.0. Anal. Calcd. for C22H15NO: C, 85.40; N, 4.53; H, 4.89; found C, 84.98; N, 4.32; H, 4.62. HRMS calcd. for C22H15NO: 309.1154; found 309.1143.
![[Graphic 35]](/bjoc/content/inline/1860-5397-5-35-i35.svg?max-width=637&scale=1.18182)
[6-(3-Fluorophenyl)quinolin-8-yl](phenyl)methanone (4g). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-nicotinaldehyde (1f) and 1-ethynyl-3-fluorobenzene were reacted and 750 μL of the crude reaction mixture were collected. Purification by flash chromatography (25% ethyl acetate in pentane) afforded 63.8 mg of 4g as a colourless oil (54% yield). 1H NMR (400 MHz, CDCl3): δ 8.87 (dd, J = 4.0, 1.5 Hz, 1H), 8.29 (dd, J = 8.1, 1.5 Hz, 1H), 8.15 (d, J = 2.0 Hz, 1H), 7.99 (d, J = 2.0 Hz, 1H), 7.90 (dd, J = 8.1, 1.5 Hz, 2H), 7.60 (t, J = 7.1 Hz, 1H), 7.40–7.56 (m, 6H), 7.10–7.17 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 197.6, 163.3 (1J13C-19F = 245.0 Hz), 151.1, 145.7, 141.8, 141.7, 140.1, 137.6, 137.5, 136.2, 133.4, 130.6 (3J13C-19F = 8.5 Hz), 130.2, 128.4, 127.5, 127.3, 123.1, 122.1, 114.9 (2J13C-19F = 22.0 Hz), 114.4 (2J13C-19F = 23.0 Hz). Anal. Calcd. for C22H14FNO: C, 80.72; N, 4.28; H, 4.31; found C, 80.77; N, 4.56; H, 4.09. HRMS calcd. for C22H14FNO: 327.1059; found 327.1046.
![[Graphic 36]](/bjoc/content/inline/1860-5397-5-35-i36.svg?max-width=637&scale=1.18182)
Phenyl-[6-(trimethylsilyl)quinolin-8-yl]methanone (4h). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-nicotinaldehyde (1f) and TMS acetylene were reacted and 800 μL of the crude reaction mixture were collected. Purification by flash chromatography (20% ethyl acetate in pentane) afforded 70.0 mg of 4h as a pale-brown oil (58% yield). 1H NMR (400 MHz, CDCl3): δ 8.82 (dd, J = 4.1, 1.5 Hz, 1H), 8.21 (dd, J = 8.1, 1.5 Hz, 1H), 8.11 (d, J = 1.6 Hz, 1H), 7.84–7.87 (m, 3H), 7.55 (t, J = 7.6 Hz, 1H), 7.38–7.44 (m, 3H), 0.38 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 198.5, 151.0, 146.4, 138.9, 138.3, 137.9, 136.0, 135.5, 133.2, 132.1, 130.2, 128.3, 127.5, 121.7, 0.87. Anal. Calcd. for C19H19NOSi: C, 74.71; N, 4.59; H, 6.27; found C, 74.93; N, 4.62; H, 6.07. HRMS calcd. for C19H19NOSi: 305.1236; found 305.1232.
![[Graphic 37]](/bjoc/content/inline/1860-5397-5-35-i37.svg?max-width=637&scale=1.18182)
[6-(4-Bromophenyl)quinolin-8-yl]-phenylmethanone (4i). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-nicotinaldehyde (1f) and 1-bromo-4-ethynylbenzene were reacted and 700 μL of the crude reaction mixture were collected. Purification by flash chromatography (20% ethyl acetate in pentane) afforded 70.0 mg of 4i as a colourless oil (52% yield). 1H NMR (400 MHz, CD2Cl2): δ 8.83 (dd, J = 4.1, 1.5 Hz, 1H), 8.35 (dd, J = 8.1, 1.5 Hz, 1H), 8.21 (d, J = 2.0 Hz, 1H), 8.00 (d, J = 2.0 Hz, 1H), 7.85 (dd, J = 8.1, 1.5 Hz, 2H), 7.60–7.76 (m, 4H), 7.44–7.56 (m, 3H), 7.34 (q, J = 9.0 Hz, 1H). 13C NMR (150 MHz, CD2Cl2): δ 197.3, 150.7, 145.4, 140.0, 138.5, 137.8, 137.5, 136.2, 133.2, 132.1, 129.8, 128.9, 128.4, 128.3, 127.2, 126.9, 122.3, 122.1. HRMS calcd. for C22H14BrNO [M+H]+: 388.0337; found 388.0337.
![[Graphic 38]](/bjoc/content/inline/1860-5397-5-35-i38.svg?max-width=637&scale=1.18182)
3-(8-benzoylquinolin-6-yl)-N,N-diisopropylbenzamide (4j). Following the general MACOS benzannulation protocol, 2-(2-phenylethynyl)-nicotinaldehyde (1f) and 3-ethynyl-N,N-diisopropylbenzamide were reacted and 840 μL of the crude reaction mixture were collected. Purification by flash chromatography (30% ethyl acetate in pentane) afforded 72.0 mg of 4j as a colourless oil (40% yield). 1H NMR (600 MHz, CD2Cl2): δ 8.81 (dd, J = 3.8, 1.6 Hz, 1H), 8.35 (dd, J = 8.1, 1.6 Hz, 1H), 8.25 (d, J = 2.1 Hz, 1H), 8.03 (d, J = 2.1 Hz, 1H), 7.84 (dd, J = 8.1, 1.6 Hz, 2H), 7.79 (d, J = 8.1 Hz, 1H), 7.70 (s, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.44–7.52 (m, 3H), 7.35 (d, J = 8.1 Hz, 1H), 3.90 (br s, 1H), 3.54 (br s, 1H), 1.55 (s, 6H), 1.16 (s, 6H). 13C NMR (150 MHz, CD2Cl2 10 °C): δ 197.6, 170.2, 150.7, 145.5, 140.2, 140.0, 139.9, 138.1, 137.8, 136.3, 133.3, 129.9, 129.2, 128.5, 128.4, 127.5, 127.4, 127.2, 124.9, 124.6, 122.2, 51.0, 45.7, 20.4. HRMS calcd. for C29H28N2O2: 436.2151; found 436.2147.
Supporting Information
Spectra for compounds made in this manuscript are available as supporting information.
| Supporting Information File 1: NMR spectra of compounds 1d, 2j, 3c, 3d, 3f, 4c and 4e–j. | ||
| Format: PDF | Size: 8.2 MB | Download |
Acknowledgements
This work was funded by the Ontario Research and Development Challenge Fund (ORDCF), NSERC (Canada), Dalton Pharma Inc., Bruker Biospin Inc., and York University. The authors are grateful to Biotage Inc. for the donation of a Smith Creator Synthesizer™ and to Syrris Inc. for the donation of the components of an AFRICA microreactor flow system to develop this new methodology. We acknowledge Ross Davidson at Surface Science Western, University of Western Ontario, London, ON for the SEM and EDX measurements.
References
-
Tierney, P. J.; Lidstrom, P. Microwave Assisted Organic Synthesis; Blackwell Publishing: Oxford, 2005.
Return to citation in text: [1] -
Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. doi:10.1002/anie.200400655
Return to citation in text: [1] -
Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577
Return to citation in text: [1] -
Pennemann, H.; Watts, P.; Haswell, S. J.; Hessel, V.; Löwe, H. Org. Process Res. Dev. 2004, 8, 422–439. doi:10.1021/op0341770
Return to citation in text: [1] -
Bremner, S.; Organ, M. G. J. Comb. Chem. 2007, 9, 14–16. doi:10.1021/cc060130p
Return to citation in text: [1] [2] -
Comer, E.; Organ, M. G. J. Am. Chem. Soc. 2005, 127, 8160–8167. doi:10.1021/ja0512069
Return to citation in text: [1] [2] -
Comer, E.; Organ, M. G. Chem.–Eur. J. 2005, 11, 7223–7227. doi:10.1002/chem.200500820
Return to citation in text: [1] [2] [3] -
Bagley, M. C.; Jenkins, R. L.; Lubinu, M. C.; Mason, C.; Wood, R. J. Org. Chem. 2005, 70, 7003–7006. doi:10.1021/jo0510235
Return to citation in text: [1] [2] -
Saaby, S.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3365–3368. doi:10.1039/b509540a
Return to citation in text: [1] [2] -
He, P.; Haswell, S. J.; Fletcher, P. D. I. Appl. Catal., A 2004, 274, 111–114.
Return to citation in text: [1] [2] -
Cablewski, T.; Faux, A. F.; Strauss, C. R. J. Org. Chem. 1994, 59, 3408–3412. doi:10.1021/jo00091a033
Return to citation in text: [1] [2] -
Glasnov, T. N.; Kappe, C. O. Macromol. Rapid Commun. 2007, 28, 395–410. doi:10.1002/marc.200600665
Return to citation in text: [1] [2] -
Glasnov, T. N.; Vugts, D. J.; Koningstein, M. M.; Desai, B.; Fabian, W. M. F.; Orru, R. V. A.; Kappe, C. O. QSAR Comb. Sci. 2006, 25, 509–518. doi:10.1002/qsar.200540210
Return to citation in text: [1] [2] -
Wilson, N. S.; Sarko, C. R.; Roth, G. Org. Process Res. Dev. 2004, 8, 535–538. doi:10.1021/op034181b
Return to citation in text: [1] [2] -
Wiles, C.; Watts, P. Eur. J. Org. Chem. 2008, 10, 1655–1671. doi:10.1002/ejoc.200701041
Return to citation in text: [1] [2] -
Wheeler, R. C.; Benali, O.; Deal, M.; Farrant, E.; MacDonald, S. J. F.; Warrington, B. H. Org. Process Res. Dev. 2007, 11, 704–710. doi:10.1021/op7000707
Return to citation in text: [1] [2] -
Baxendale, I. R.; Hayward, J. J.; Ley, S. V. Comb. Chem. High Throughput Screening 2007, 10, 802–836. doi:10.2174/138620707783220374
Return to citation in text: [1] [2] -
Brivio, M.; Verboom, W.; Reinhoudt, D. N. Lab Chip 2006, 6, 329–344. doi:10.1039/b510856j
Return to citation in text: [1] [2] -
Wild, G. P.; Wiles, C.; Watts, P. Lett. Org. Chem. 2006, 3, 419–425. doi:10.2174/157017806777828475
Return to citation in text: [1] [2] -
Watts, P.; Haswell, S. J. Drug Discovery Today 2003, 8, 586–593. doi:10.1016/S1359-6446(03)02732-6
Return to citation in text: [1] [2] -
Jones, R. V.; Csajagi, C.; Szekelyhidi, Z.; Kovacs, I.; Borcsek, B.; Urge, L.; Darvas, F. Chim. Oggi 2008, 26 (3), 10–12.
Return to citation in text: [1] [2] -
Kikutani, Y.; Kitamori, T. Macromol. Rapid Commun. 2004, 25, 158–168. doi:10.1002/marc.200300192
Return to citation in text: [1] [2] -
Stonestreet, P.; Harvey, A. P. Chem. Eng. Res. Des. 2002, 80, 31–44. doi:10.1205/026387602753393204
Return to citation in text: [1] [2] -
Vasudevan, D.; Basha, C. A. Bull. Electrochem. 2000, 16, 341–344.
Return to citation in text: [1] [2] -
Shore, G.; Organ, M. G. Chem.–Eur. J. 2008, 14, 9641–9646. doi:10.1002/chem.200801610
Return to citation in text: [1] [2] -
Shore, G.; Organ, M. G. Chem. Commun. 2008, 838–840. doi:10.1039/b715709f
Return to citation in text: [1] -
Shore, G.; Morin, S.; Mallik, D.; Organ, M. G. Chem.–Eur. J. 2008, 14, 1351–1356. doi:10.1002/chem.200701588
Return to citation in text: [1] -
Shore, G.; Morin, S.; Organ, M. G. Angew. Chem., Int. Ed. 2006, 45, 2761–2766. doi:10.1002/anie.200503600
Return to citation in text: [1] [2] -
Hashmi, A. S. K. Gold Bull. 2003, 36, 3–9.
Return to citation in text: [1] -
Asao, N. Synlett 2006, 1645–1656. doi:10.1055/s-2006-947331
Return to citation in text: [1] -
Asao, N.; Nogami, T.; Lee, S.; Yamamoto, Y. J. Am. Chem. Soc. 2003, 125, 10921–10925. doi:10.1021/ja036927r
Return to citation in text: [1] -
Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d
Return to citation in text: [1] -
Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. Org. Lett. 2001, 3, 3769–3771. doi:10.1021/ol016734d
Return to citation in text: [1] -
Asao, N.; Takahashi, K.; Lee, S.; Kasahara, T.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 12650–12651. doi:10.1021/ja028128z
Return to citation in text: [1] [2] [3] -
Straub, B. F. Chem. Commun. 2004, 1726–1728. doi:10.1039/b404876h
Return to citation in text: [1] -
Obika, S.; Kono, H.; Yasui, R.; Yanada, Y.; Takemoto, Y. J. Org. Chem. 2007, 72, 4462–4468. doi:10.1021/jo070615f
Return to citation in text: [1] -
Dai, G.; Larock, R. C. Org. Lett. 2001, 3, 4035–4038. doi:10.1021/ol0102085
Return to citation in text: [1] -
Dyker, G.; Stirner, W.; Henkel, G. Eur. J. Org. Chem. 2000, 8, 1433–1441. doi:10.1002/(SICI)1099-0690(200004)2000:8<1433::AID-EJOC1433>3.0.CO;2-7
Return to citation in text: [1] -
Barluenga, J.; Vasquez-Villa, H.; Ballesteros, A.; Gonzalez, J. M. Org. Lett. 2003, 5, 4121–4124. doi:10.1021/ol035691t
Return to citation in text: [1] -
Asao, N.; Aikawa, A.; Yamammoto, Y. J. Am. Chem. Soc. 2004, 126, 7458–7459. doi:10.1021/ja0477367
Return to citation in text: [1]
| 34. | Asao, N.; Takahashi, K.; Lee, S.; Kasahara, T.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 12650–12651. doi:10.1021/ja028128z |
| 40. | Asao, N.; Aikawa, A.; Yamammoto, Y. J. Am. Chem. Soc. 2004, 126, 7458–7459. doi:10.1021/ja0477367 |
| 34. | Asao, N.; Takahashi, K.; Lee, S.; Kasahara, T.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 12650–12651. doi:10.1021/ja028128z |
| 1. | Tierney, P. J.; Lidstrom, P. Microwave Assisted Organic Synthesis; Blackwell Publishing: Oxford, 2005. |
| 2. | Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. doi:10.1002/anie.200400655 |
| 25. | Shore, G.; Organ, M. G. Chem.–Eur. J. 2008, 14, 9641–9646. doi:10.1002/chem.200801610 |
| 26. | Shore, G.; Organ, M. G. Chem. Commun. 2008, 838–840. doi:10.1039/b715709f |
| 27. | Shore, G.; Morin, S.; Mallik, D.; Organ, M. G. Chem.–Eur. J. 2008, 14, 1351–1356. doi:10.1002/chem.200701588 |
| 28. | Shore, G.; Morin, S.; Organ, M. G. Angew. Chem., Int. Ed. 2006, 45, 2761–2766. doi:10.1002/anie.200503600 |
| 38. | Dyker, G.; Stirner, W.; Henkel, G. Eur. J. Org. Chem. 2000, 8, 1433–1441. doi:10.1002/(SICI)1099-0690(200004)2000:8<1433::AID-EJOC1433>3.0.CO;2-7 |
| 5. | Bremner, S.; Organ, M. G. J. Comb. Chem. 2007, 9, 14–16. doi:10.1021/cc060130p |
| 6. | Comer, E.; Organ, M. G. J. Am. Chem. Soc. 2005, 127, 8160–8167. doi:10.1021/ja0512069 |
| 7. | Comer, E.; Organ, M. G. Chem.–Eur. J. 2005, 11, 7223–7227. doi:10.1002/chem.200500820 |
| 8. | Bagley, M. C.; Jenkins, R. L.; Lubinu, M. C.; Mason, C.; Wood, R. J. Org. Chem. 2005, 70, 7003–7006. doi:10.1021/jo0510235 |
| 9. | Saaby, S.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3365–3368. doi:10.1039/b509540a |
| 10. | He, P.; Haswell, S. J.; Fletcher, P. D. I. Appl. Catal., A 2004, 274, 111–114. |
| 11. | Cablewski, T.; Faux, A. F.; Strauss, C. R. J. Org. Chem. 1994, 59, 3408–3412. doi:10.1021/jo00091a033 |
| 12. | Glasnov, T. N.; Kappe, C. O. Macromol. Rapid Commun. 2007, 28, 395–410. doi:10.1002/marc.200600665 |
| 13. | Glasnov, T. N.; Vugts, D. J.; Koningstein, M. M.; Desai, B.; Fabian, W. M. F.; Orru, R. V. A.; Kappe, C. O. QSAR Comb. Sci. 2006, 25, 509–518. doi:10.1002/qsar.200540210 |
| 14. | Wilson, N. S.; Sarko, C. R.; Roth, G. Org. Process Res. Dev. 2004, 8, 535–538. doi:10.1021/op034181b |
| 15. | Wiles, C.; Watts, P. Eur. J. Org. Chem. 2008, 10, 1655–1671. doi:10.1002/ejoc.200701041 |
| 16. | Wheeler, R. C.; Benali, O.; Deal, M.; Farrant, E.; MacDonald, S. J. F.; Warrington, B. H. Org. Process Res. Dev. 2007, 11, 704–710. doi:10.1021/op7000707 |
| 17. | Baxendale, I. R.; Hayward, J. J.; Ley, S. V. Comb. Chem. High Throughput Screening 2007, 10, 802–836. doi:10.2174/138620707783220374 |
| 18. | Brivio, M.; Verboom, W.; Reinhoudt, D. N. Lab Chip 2006, 6, 329–344. doi:10.1039/b510856j |
| 19. | Wild, G. P.; Wiles, C.; Watts, P. Lett. Org. Chem. 2006, 3, 419–425. doi:10.2174/157017806777828475 |
| 20. | Watts, P.; Haswell, S. J. Drug Discovery Today 2003, 8, 586–593. doi:10.1016/S1359-6446(03)02732-6 |
| 21. | Jones, R. V.; Csajagi, C.; Szekelyhidi, Z.; Kovacs, I.; Borcsek, B.; Urge, L.; Darvas, F. Chim. Oggi 2008, 26 (3), 10–12. |
| 22. | Kikutani, Y.; Kitamori, T. Macromol. Rapid Commun. 2004, 25, 158–168. doi:10.1002/marc.200300192 |
| 23. | Stonestreet, P.; Harvey, A. P. Chem. Eng. Res. Des. 2002, 80, 31–44. doi:10.1205/026387602753393204 |
| 24. | Vasudevan, D.; Basha, C. A. Bull. Electrochem. 2000, 16, 341–344. |
| 39. | Barluenga, J.; Vasquez-Villa, H.; Ballesteros, A.; Gonzalez, J. M. Org. Lett. 2003, 5, 4121–4124. doi:10.1021/ol035691t |
| 5. | Bremner, S.; Organ, M. G. J. Comb. Chem. 2007, 9, 14–16. doi:10.1021/cc060130p |
| 6. | Comer, E.; Organ, M. G. J. Am. Chem. Soc. 2005, 127, 8160–8167. doi:10.1021/ja0512069 |
| 7. | Comer, E.; Organ, M. G. Chem.–Eur. J. 2005, 11, 7223–7227. doi:10.1002/chem.200500820 |
| 8. | Bagley, M. C.; Jenkins, R. L.; Lubinu, M. C.; Mason, C.; Wood, R. J. Org. Chem. 2005, 70, 7003–7006. doi:10.1021/jo0510235 |
| 9. | Saaby, S.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3365–3368. doi:10.1039/b509540a |
| 10. | He, P.; Haswell, S. J.; Fletcher, P. D. I. Appl. Catal., A 2004, 274, 111–114. |
| 11. | Cablewski, T.; Faux, A. F.; Strauss, C. R. J. Org. Chem. 1994, 59, 3408–3412. doi:10.1021/jo00091a033 |
| 12. | Glasnov, T. N.; Kappe, C. O. Macromol. Rapid Commun. 2007, 28, 395–410. doi:10.1002/marc.200600665 |
| 13. | Glasnov, T. N.; Vugts, D. J.; Koningstein, M. M.; Desai, B.; Fabian, W. M. F.; Orru, R. V. A.; Kappe, C. O. QSAR Comb. Sci. 2006, 25, 509–518. doi:10.1002/qsar.200540210 |
| 14. | Wilson, N. S.; Sarko, C. R.; Roth, G. Org. Process Res. Dev. 2004, 8, 535–538. doi:10.1021/op034181b |
| 15. | Wiles, C.; Watts, P. Eur. J. Org. Chem. 2008, 10, 1655–1671. doi:10.1002/ejoc.200701041 |
| 16. | Wheeler, R. C.; Benali, O.; Deal, M.; Farrant, E.; MacDonald, S. J. F.; Warrington, B. H. Org. Process Res. Dev. 2007, 11, 704–710. doi:10.1021/op7000707 |
| 17. | Baxendale, I. R.; Hayward, J. J.; Ley, S. V. Comb. Chem. High Throughput Screening 2007, 10, 802–836. doi:10.2174/138620707783220374 |
| 18. | Brivio, M.; Verboom, W.; Reinhoudt, D. N. Lab Chip 2006, 6, 329–344. doi:10.1039/b510856j |
| 19. | Wild, G. P.; Wiles, C.; Watts, P. Lett. Org. Chem. 2006, 3, 419–425. doi:10.2174/157017806777828475 |
| 20. | Watts, P.; Haswell, S. J. Drug Discovery Today 2003, 8, 586–593. doi:10.1016/S1359-6446(03)02732-6 |
| 21. | Jones, R. V.; Csajagi, C.; Szekelyhidi, Z.; Kovacs, I.; Borcsek, B.; Urge, L.; Darvas, F. Chim. Oggi 2008, 26 (3), 10–12. |
| 22. | Kikutani, Y.; Kitamori, T. Macromol. Rapid Commun. 2004, 25, 158–168. doi:10.1002/marc.200300192 |
| 23. | Stonestreet, P.; Harvey, A. P. Chem. Eng. Res. Des. 2002, 80, 31–44. doi:10.1205/026387602753393204 |
| 24. | Vasudevan, D.; Basha, C. A. Bull. Electrochem. 2000, 16, 341–344. |
| 36. | Obika, S.; Kono, H.; Yasui, R.; Yanada, Y.; Takemoto, Y. J. Org. Chem. 2007, 72, 4462–4468. doi:10.1021/jo070615f |
| 3. | Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577 |
| 4. | Pennemann, H.; Watts, P.; Haswell, S. J.; Hessel, V.; Löwe, H. Org. Process Res. Dev. 2004, 8, 422–439. doi:10.1021/op0341770 |
| 31. | Asao, N.; Nogami, T.; Lee, S.; Yamamoto, Y. J. Am. Chem. Soc. 2003, 125, 10921–10925. doi:10.1021/ja036927r |
| 29. | Hashmi, A. S. K. Gold Bull. 2003, 36, 3–9. |
| 30. | Asao, N. Synlett 2006, 1645–1656. doi:10.1055/s-2006-947331 |
| 7. | Comer, E.; Organ, M. G. Chem.–Eur. J. 2005, 11, 7223–7227. doi:10.1002/chem.200500820 |
| 25. | Shore, G.; Organ, M. G. Chem.–Eur. J. 2008, 14, 9641–9646. doi:10.1002/chem.200801610 |
| 28. | Shore, G.; Morin, S.; Organ, M. G. Angew. Chem., Int. Ed. 2006, 45, 2761–2766. doi:10.1002/anie.200503600 |
| 32. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d |
| 33. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. Org. Lett. 2001, 3, 3769–3771. doi:10.1021/ol016734d |
| 34. | Asao, N.; Takahashi, K.; Lee, S.; Kasahara, T.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 12650–12651. doi:10.1021/ja028128z |
© 2009 Shore et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)