Abstract
The reactions of benzo[e][2,1]thiazine-4-chloro-3-carbaldehydes 1 and benzo[e][2,1]thiazine-4-chloro-3-carbonitriles 2 with a number of oxidizing and reducing agents are reported. A number of new, highly functionalized benzo[e][2,1]thiazine derivatives having potential biological activity were synthesized and described.

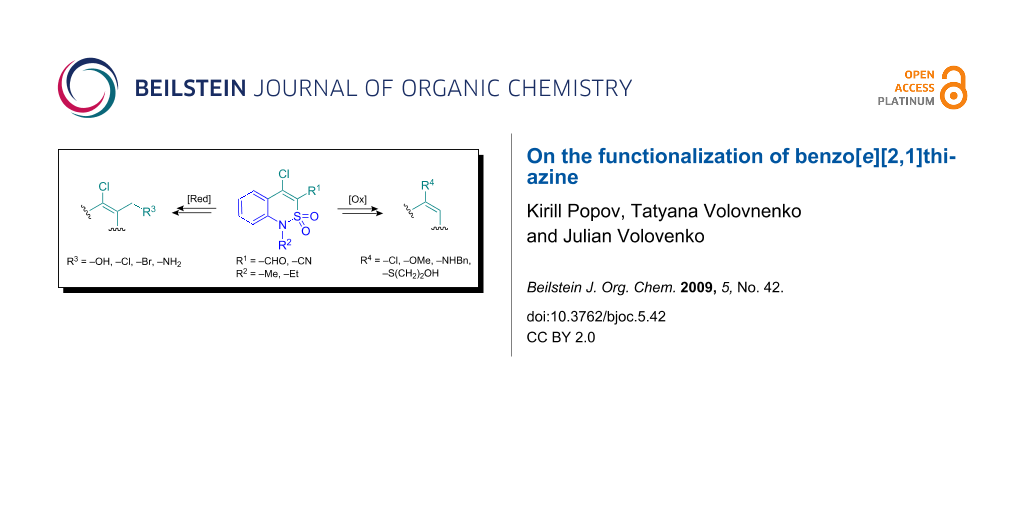
Graphical Abstract
Introduction
Many derivatives of benzothiazine exhibit biological activity that ranges from antipsychotic [1] to anti-inflammatory [2], depending on the substituents present [3,4]. However, medicinal applications are limited as a consequence of side effects [5,6]. Much research has been carried out over the last 20 years with a view to improving benzothiazine-based drugs and to avoid the adverse effects [6,7].
In previous work [8,9] we reported the synthesis of some novel benzothiazines, in particular benzo[e][2,1]thiazine derivatives, the β-chloroaldehydes 1 and β-chloronitriles 2 (Figure 1); their chemistry was shown to be quite versatile. Compounds 1 and 2 both contain 1,3-dielectrophilic fragments, which are C-4 carbon atoms of the benzothiazine ring and carbonyl group or nitrile function, respectively. Thus, further investigation of aldehydes 1 and nitriles 2 should provide new benzo[e][2,1]thiazine derivatives, which can be used as intermediates for the synthesis of more elaborate compounds with potential biological activity.
![[1860-5397-5-42-1]](/bjoc/content/figures/1860-5397-5-42-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Benzo[e][2,1]thiazine-4-chloro-3-carbaldehydes 1 and benzo[e][2,1]thiazine-4-chloro-3-carbonitriles 2.
Figure 1: Benzo[e][2,1]thiazine-4-chloro-3-carbaldehydes 1 and benzo[e][2,1]thiazine-4-chloro-3-carbonitriles ...
The current article describes the chemical behaviour of chloroaldehydes 1a,b and chloronitriles 2a,b (Table 1) in oxidation and reduction reactions.
Results and Discussion
Chloroaldehydes 1a,b are readily reduced under mild conditions by sodium borohydride to yield the alcohols 3a,b. Treatment of compounds 3a,b with thionyl chloride in dry benzene results in the formation of dichloro derivatives 4a,b, whilst the 3-bromomethyl derivatives 5a,b are obtained by refluxing 3a,b in concentrated hydrobromic acid. Nucleophilic substitution of the chlorine atoms in compounds 4 shows similar behaviour. Thus, treatment of the dichloro derivatives 4a,b with sodium methoxide gives a mixture of substitution products in a 2:1 isomer ratio with side-chain substitution predominating. The bromine atom in compounds 5 is much more reactive than the chlorines in 4. Thus, when 5a,b were heated with 2-mercaptoethanol and triethylamine in dioxane, compounds 6a,b were obtained as the sole products (Scheme 1).
![[1860-5397-5-42-i1]](/bjoc/content/inline/1860-5397-5-42-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: a: NaBH4 (2.5 equiv), MeOH, 2 h, room temp.; b: SOCl2 (4 equiv), benzene, 3 h, 0 °C to room temp.; c: HBr (50%), 5 h, reflux; d: NaOMe/MeOH, 2 h, reflux; e: HS–(CH2)2–OH (1.5 equiv), K2CO3, 1,4-dioxane, TEA, 2 h, reflux.
Scheme 1: a: NaBH4 (2.5 equiv), MeOH, 2 h, room temp.; b: SOCl2 (4 equiv), benzene, 3 h, 0 °C to room temp.; c...
Amines 7a,b were synthesized by reduction of chloronitriles 2a,b with lithium aluminium hydride in dry diethyl ether or THF. Compounds 7 are very unstable. Presumably, intermolecular arylation of the active amino group occurs rapidly. Consequently, amines 7a,b were isolated as their hydrochlorides (Scheme 2).
![[1860-5397-5-42-i2]](/bjoc/content/inline/1860-5397-5-42-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: a: LiAlH4 (4 equiv), Et2O (THF), 3 h, 0 °C to room temp.; b: HCl saturated in 1,4-dioxane.
Scheme 2: a: LiAlH4 (4 equiv), Et2O (THF), 3 h, 0 °C to room temp.; b: HCl saturated in 1,4-dioxane.
Despite our expectations, aldehydes 1 appeared to be inert to the most common oxidizing agents. Thus, oxidants such as sodium dichromate and potassium permanganate did not transform 1 into the corresponding carboxylic acids. Attempted oxidation of compounds 1 with hydrogen peroxide, peroxyacetic acid and m-chloroperoxybenzoic acid (mCPBA) was also unsuccessful. The desired oxidation reaction occurs with silver(I) oxide, prepared in situ from silver nitrate. Aldehydes 1a,b are oxidized under mild conditions; however, the resulting carboxylic acids 8 could not be isolated, since even at low temperatures (up to 3 °C) and with immediate adjustment of the pH (between pH = 10 and pH = 3) instantaneous decarboxylation was observed, and 4-chlorobenzo[e][2,1]thiazines 9a,b were obtained in quantitative yield. The chlorine atom in compounds 9a,b is readily substituted by O-, N- and S-nucleophiles. Reaction of 9a,b with sodium methoxide, benzylamine, and 2-mercaptoethanol gave the corresponding 4-methoxybenzothiazines 10a,b, N-benzylamines 11a,b and sulfanylethanol derivatives 12a,b, respectively (Scheme 3).
![[1860-5397-5-42-i3]](/bjoc/content/inline/1860-5397-5-42-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: a: AgNO3 (1.5 equiv), NaOH, H2O/CH2Cl2, 3 h, room temp.; b: NaOMe/MeOH, 1 h, reflux; c: PhCH2NH2 (2 equiv), i-PrOH, 2 h, reflux; d: HS–(CH2)2–OH (1.5 equiv), K2CO3, dioxane, TEA, 1 h, reflux.
Scheme 3: a: AgNO3 (1.5 equiv), NaOH, H2O/CH2Cl2, 3 h, room temp.; b: NaOMe/MeOH, 1 h, reflux; c: PhCH2NH2 (2...
Conclusion
In summary, we describe the preparation of novel benzo[e][2,1]thiazine derivatives capable of further modification. In particular β-hydroxymethylchlorides 3a,b, amines 7a,b and 4-chlorobenzo[e][2,1]thiazines 9a,b were obtained in high yields (95–99%) via convenient protocols involving oxidation and reduction reactions of compounds 1 and 2. Biological testing of the synthesized compounds is currently in progress. The structures of all the described products (Table 2) were established from their NMR spectra, elemental analyses and mass spectra.
Experimental
General procedure for the synthesis of compounds 3
To a magnetically stirred suspension of 1a,b (1 g, 4 mmol) in dry methanol (5 mL) sodium borohydride (0.64 g, 16 mmol) was added in small portions at room temperature. The solution was stirred for 2 hours and concentrated in vacuo. Dilute hydrochloric acid (2N HCl, 5 mL) was added, the solid product filtered off and washed with water to give pure compound 3a,b (93–95%).
General procedure for the synthesis of compounds 4
To a magnetically stirred suspension of 3a,b (1 g, 3.6 mmol) in dry benzene (6 mL) thionyl chloride (1.5 mL) was added dropwise at room temperature. The solution was stirred for 3 hours and concentrated in vacuo. Water was added and the solid product filtered off. Pure 4a,b was obtained by crystallization from hexane (95–98%).
General procedure for the synthesis of compounds 5
The mixture of 3a,b (1 g, 3.6 mmol) and concentrated hydrobromic acid (3 mL) was refluxed for 5 hours. The reaction mixture was cooled to 0 °C, the solids filtered off and washed with water to give pure compound 5a,b (71–76%).
General procedure for the synthesis of compounds 6
The mixture of compound 5a,b (1 g, 2.6 mmol), mercaptoethanol (0.5 mL), potassium carbonate (0.5 g) and triethylamine (1.5 mL) in 1,4-dioxane (5 mL) was refluxed for 2 hours, then concentrated in vacuo to afford the crude product as a brown oil. The pure product 6a,b was obtained by column chromatography (CHCl3/CH3OH 9:1, 55–57%).
General procedure for the synthesis of compounds 7
To a magnetically stirred suspension of 2a,b (1 g, 4 mmol) in dry ether (3 mL) or dry THF (3 mL) lithium aluminium hydride (0.7 g, 16 mmol) was added in small portions at 0 °C. The solution was stirred for 3 hours and the temperature gradually raised to 25 °C. The solvent was evaporated and water added to the residue. The product was extracted with CH2Cl2 (3 × 5 mL), concentrated in vacuo and treated with dry 1,4-dioxane saturated with HCl to afford the pure crystalline product 7a,b (67–68%).
General procedure for the synthesis of compounds 9
To a magnetically stirred aqueous solution of silver nitrate (1 g, 6 mmol) sodium hydroxide (0.3 g, 7.5 mmol) was added in small portions at room temperature. To the resulting suspension of Ag2O a solution of 1a,b (1 g, 4 mmol) in CH2Cl2 (3 mL) was added dropwise and the resulting mixture stirred for 3 hours. The organic layer was separated and concentrated in vacuo to give the pure solid product 9a,b (99%).
General procedure for the synthesis of compounds 10
The mixture of 9a,b (1 g, 4 mmol) and MeONa (0.5 g, 8.5 mmol) in dry methanol (5 mL) was refluxed for 1 hour. The solvent was evaporated and the solid product washed with i-PrOH to give pure compound 10a,b (85–87%).
General procedure for the synthesis of compounds 11
The mixture of 9a,b (1 g, 4 mmol) and benzylamine (0.5 g, 5 mmol) in i-PrOH (5 mL) was refluxed for 2 hours. The solvent was evaporated, and the solid product washed with i-PrOH and water. The pure product 11a,b was obtained by crystallization from toluene (60–63%).
General procedure for the synthesis of compounds 12
The mixture of 9a,b (1 g, 4 mmol), mercaptoethanol (0.5 mL), potassium carbonate (0.5 g) and triethylamine (1.5 mL) in 1,4-dioxane (7 mL) was refluxed for 1 hour. Inorganic material was removed by filtration and the resulting solution concentrated in vacuo. The oily product was purified by column chromatography on silica gel with chloroform-methanol (8:2) as eluent to give the pure product 12a,b (32–34%).
Supporting Information
Supporting information contains melting points, elemental analyses, 1H-, 13C-NMR and mass spectra data for all new compounds (3–12).
| Supporting Information File 1: Spectroscopic data for: On the functionalization of benzo[e][2,1]thiazine. | ||
| Format: DOC | Size: 60.0 KB | Download |
References
-
Yeung, P.; Hubbard, J.; Korchinski, E.; Midha, K. Eur. J. Clin. Pharmacol. 1993, 45, 563–569. doi:10.1007/BF00315316
Return to citation in text: [1] -
Olkkola, K. T.; Brunetto, A. V.; Mattila, M. J. Clin. Pharmacokinet. 1994, 26, 107–120. doi:10.2165/00003088-199426020-00004
Return to citation in text: [1] -
Catsoulacos, P.; Camoutsis, C. J. Heterocycl. Chem. 1979, 16, 1503–1524. doi:10.1002/jhet.5570160801
Return to citation in text: [1] -
Banerjee, R.; Chakraborty, H.; Sarkar, M. Spectrochim. Acta, Part A 2003, 56, 1213–1222. doi:10.1016/S1386-1425(02)00300-1
Return to citation in text: [1] -
Rossi, S., Ed. Australian Medicines Handbook 2006; Australian Medicines Handbook Pty Ltd: Adelaide, 2006.
Return to citation in text: [1] -
Van Hecken, A.; Schwartz, J. I.; Depre, M.; De Lepeleire, I.; Dallob, A.; Tanaka, W.; Wynants, K.; Buntinx, A.; Arnout, J.; Wong, P. H.; Ebel, D. L.; Gertz, B. J.; De Schepper, P. J. J. Clin. Pharmacol. 2000, 40, 1109–1120.
Return to citation in text: [1] [2] -
Chan, C.-C.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Ford-Hutchinson, A. W.; Forrest, M. J.; Gauthier, J. Y.; Gordon, R.; Gresser, M.; Guay, J.; Kargman, S.; Kennedy, B.; Leblanc, Y.; Leger, S.; Mancini, J.; O’Neill, G. P.; Ouellet, M.; Patrick, D.; Percival, M. D.; Perrier, H.; Prasit, P.; Rodger, I.; Tagari, P.; Therien, M.; Vickers, P.; Visco, D.; Wang, Z.; Webb, J.; Wong, E.; Xu, L.-J.; Young, R. N.; Zamboni, R.; Riendeau, D. Pharmacol. Exp. Ther. 1999, 290, 551–560.
Return to citation in text: [1] -
Volovenko, Y. M.; Volovnenko, T. A.; Popov, K. S. J. Heterocycl. Chem. 2007, 44, 1413–1420. doi:10.1002/jhet.5570440627
Return to citation in text: [1] -
Volovenko, Y. M.; Volovnenko, T. A.; Popov, K. S. Tetrahedron Lett. 2009, 50, 1171–1172. doi:10.1016/j.tetlet.2008.10.101
Return to citation in text: [1]
| 1. | Yeung, P.; Hubbard, J.; Korchinski, E.; Midha, K. Eur. J. Clin. Pharmacol. 1993, 45, 563–569. doi:10.1007/BF00315316 |
| 6. | Van Hecken, A.; Schwartz, J. I.; Depre, M.; De Lepeleire, I.; Dallob, A.; Tanaka, W.; Wynants, K.; Buntinx, A.; Arnout, J.; Wong, P. H.; Ebel, D. L.; Gertz, B. J.; De Schepper, P. J. J. Clin. Pharmacol. 2000, 40, 1109–1120. |
| 7. | Chan, C.-C.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Ford-Hutchinson, A. W.; Forrest, M. J.; Gauthier, J. Y.; Gordon, R.; Gresser, M.; Guay, J.; Kargman, S.; Kennedy, B.; Leblanc, Y.; Leger, S.; Mancini, J.; O’Neill, G. P.; Ouellet, M.; Patrick, D.; Percival, M. D.; Perrier, H.; Prasit, P.; Rodger, I.; Tagari, P.; Therien, M.; Vickers, P.; Visco, D.; Wang, Z.; Webb, J.; Wong, E.; Xu, L.-J.; Young, R. N.; Zamboni, R.; Riendeau, D. Pharmacol. Exp. Ther. 1999, 290, 551–560. |
| 5. | Rossi, S., Ed. Australian Medicines Handbook 2006; Australian Medicines Handbook Pty Ltd: Adelaide, 2006. |
| 6. | Van Hecken, A.; Schwartz, J. I.; Depre, M.; De Lepeleire, I.; Dallob, A.; Tanaka, W.; Wynants, K.; Buntinx, A.; Arnout, J.; Wong, P. H.; Ebel, D. L.; Gertz, B. J.; De Schepper, P. J. J. Clin. Pharmacol. 2000, 40, 1109–1120. |
| 3. | Catsoulacos, P.; Camoutsis, C. J. Heterocycl. Chem. 1979, 16, 1503–1524. doi:10.1002/jhet.5570160801 |
| 4. | Banerjee, R.; Chakraborty, H.; Sarkar, M. Spectrochim. Acta, Part A 2003, 56, 1213–1222. doi:10.1016/S1386-1425(02)00300-1 |
| 2. | Olkkola, K. T.; Brunetto, A. V.; Mattila, M. J. Clin. Pharmacokinet. 1994, 26, 107–120. doi:10.2165/00003088-199426020-00004 |
| 8. | Volovenko, Y. M.; Volovnenko, T. A.; Popov, K. S. J. Heterocycl. Chem. 2007, 44, 1413–1420. doi:10.1002/jhet.5570440627 |
| 9. | Volovenko, Y. M.; Volovnenko, T. A.; Popov, K. S. Tetrahedron Lett. 2009, 50, 1171–1172. doi:10.1016/j.tetlet.2008.10.101 |
© 2009 Popov et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)