Abstract
A 2-diethanolamine boronyl substituted 1,3-diene has been synthesized in high yield and characterized spectroscopically as well as by X-ray crystallography. This diene has then subsequently been used in a number of fast, high yielding Diels–Alder/cross coupling reactions.

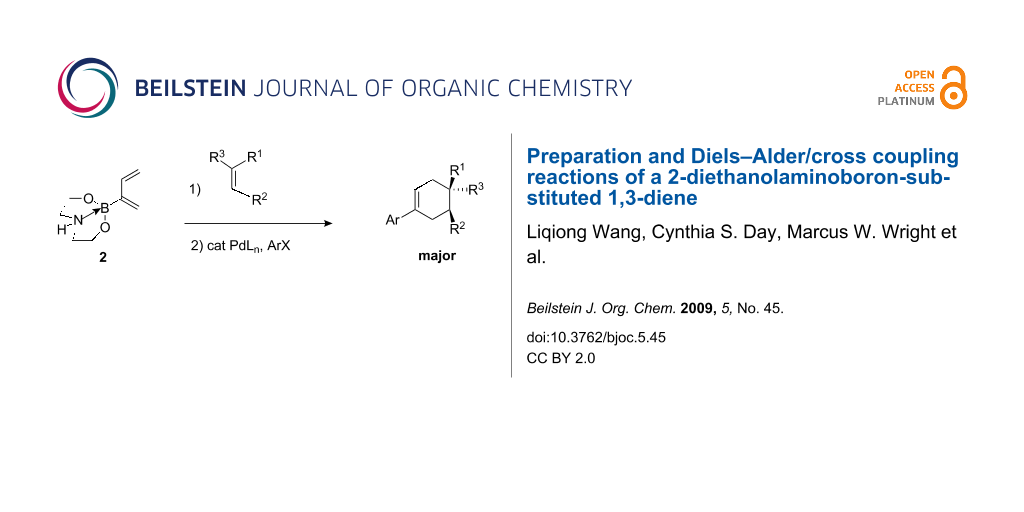
Graphical Abstract
Introduction
Our group [1] and the Tada group [2] independently reported the preparation and Diels–Alder reactions of pyridine cobaloxime dienyl complexes over 15 years ago. Since that time, we have reported a number of synthetic routes to these and other related types of cobalt dienyl complexes as well as their subsequent cycloaddition and demetallation chemistry [3-5], and other groups have now made use of the cycloadducts thus prepared [6] as well as the methodology [7].
We have now subsequently reported the preparation of 2-BF3 substituted 1,3-butadienes and demonstrated that they can be used in sequential Diels–Alder/cross coupling reactions [8,9]. These trifluoroborate substituted dienes are stable but their organic solvent solubility is not ideal. Preparation of more highly substituted BF3 dienes also requires a transmetallation protocol which yields a Grignard-BF3 by-product which has to be separated from the desired diene [9]. To overcome these methodology challenges we have begun to prepare diethanolaminoboron substituted dienes and we communicate our first results in this area here.
Results and Discussion
The diethanolamine boronyl substituted diene 2 was obtained as white needles on a several gram scale from a simple procedure which involved preparing the Grignard reagent from chloroprene 1, adding this reagent to trimethoxyborane followed by the addition of dilute HCl and diethanolamine (Scheme 1). The boron substituted diene 2 thus obtained has C1 (δ 5.23 vs δ 5.04, 4.96 (d6-DMSO) and C3 (δ 6.31 vs. δ 6.19) hydrogen atoms which are significantly more deshielded than the BF3 substituted diene. In the solid state (Figure 1, see Supporting Information), C(1)–C(2) and C(2)–C(3) bond lengths were virtually identical in both dienes whereas B–C(2) (1.609(5) Å vs 1.576(13) Å) and C(3)–C(4) (1.308(6) Å vs 1.279(13) Å) were significantly longer in the diethanolamine boronyl diene 2.
![[1860-5397-5-45-i1]](/bjoc/content/inline/1860-5397-5-45-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2-diethanolaminoborate-1,3-butadiene.
Scheme 1: Synthesis of 2-diethanolaminoborate-1,3-butadiene.
![[1860-5397-5-45-1]](/bjoc/content/figures/1860-5397-5-45-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of boron substituted diene 2.
Figure 1: Molecular structure of boron substituted diene 2.
This diethanolamine boronyl diene 2 has proved to be significantly more reactive and more regioselective in Diels–Alder reactions compared to its BF3 diene counterpart (Table 1, Scheme 2) [8]. Qualitatively, we initially noticed that whereas the BF3 diene required 16 h of heating at 95–100 °C in a sealed tube in toluene with N-phenylmaleimide to obtain >90% yield of cycloadduct, the diethanolamine boronyl diene 2 reacted with this same dienophile to afford a 98% isolated yield of cyclaooduct 4 after only 15 min at 25 °C! We tried to get more quantitative rate constant data about this Diels–Alder reaction via NMR spectroscopy but when we try to perform this reaction under pseudo first order conditions at −10 °C, we can only say that the t1/2 is less than 4 minutes. Attempts to get more accurate kinetic data by NMR at −40 °C resulted instead in diene 2 precipitation. This diene 2 is by far the most reactive main group element substituted diene we have made in the boron or silicon substituted series to date. What is perhaps even more surprising to us is that this diene 2 is even more reactive than the most reactive cobaloxime substituted diene we ever prepared in our earlier work [10] and those cobaloxime dienes consistently favored the s-cis conformation in the solid state. Diene 2 is in the s-trans conformation in the solid state (Figure 1) but in this case we suspect that the preference for the s-trans conformer is due to intermolecular hydrogen bonding between the N–H and one of the adjacent molecule’s boronate oxygen atoms. This hydrogen bonding would make a C(2)–C(3) dihedral angle of 50–60° (on the order of those we observed in cobaloxime diene solid state structures) unfavorable. At 25 °C in CDCl3, we saw no evidence for the s-cis conformer by NOESY.
![[1860-5397-5-45-i2]](/bjoc/content/inline/1860-5397-5-45-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
In an effort to further understand this reactivity difference, geometry-optimization with DFT using the B3LYP functional and a 6-31G(d) basis set followed by population analysis was performed using Gaussian 03 on 2-diethanolaminoboronyl-1,3-butadiene (2) and its BF3 diene counterpart. 2-Diethanolaminoboronyl-1,3-butadiene has a HOMO energy of −6.00 eV, whereas its BF3 diene counterpart has a HOMO energy of −12.58 eV. These energies are consistent with our observations that 2-diethanolaminoboronyl-1,3-butadiene (2) is more reactive than its BF3 diene counterpart. Furthermore, a Mulliken population analysis indicates a build-up of electron density on carbons C1 and C4 of 0.15e and 0.14e respectively in 2-diethanolaminoboronyl-1,3-butadiene (2) compared to its BF3 diene counterpart, which is also consistent with our observations. In addition to enhanced Diels–Alder reaction rates, we also noted greatly improved regioselectivities (Table 1). Whereas the BF3 diene required 36 h of heating to 95–100 °C in a sealed tube in ethanol to provide a 3.3:1 mixture of regioisomers from reaction with ethyl acrylate, the diethanolamine boronyl diene 2 reacted with this same dienophile at reflux for 6 h to provide a 16.4:1 mixture of para (1,4) to meta (1,3) isomers 3 in identical isolated yield. Similarly, when we used a citraconamide derivative (2-methyl N-phenylmaleimide), we isolated cycloadduct 5 in high yield although with reduced regioselectivity (4:1). However, the BF3 diene proved unreactive with citraconic acid derived dienophiles. This diethanolamine boronyl diene 2 once again reacted under much milder conditions and with better regioselectivity than highly reactive silicon substituted dienes we have also reported previously [11].
Lastly, in order to prove that diethanolamine boronyl diene 2 could serve as a synthon for a host of other organic dienes, we took cycloadducts 3–5 and proved that they could be cross coupled efficiently to iodobenzene, 4-trifluoromethyl-1-iodobenzene, and 4-iodoanisole (Table 2, Scheme 3). Cross coupled cycloadducts 6–14 were all isolated in good to excellent yield and regioselectivities observed in the original Diels–Alder reactions were maintained after cross coupling.
![[1860-5397-5-45-i3]](/bjoc/content/inline/1860-5397-5-45-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we report a simple preparation of a 2-boronyl substituted 1,3 diene which has proved to be the most reactive 2-main group element or 2-transition metal element substituted diene for Diels–Alder reactions that we have prepared to date. We have also demonstrated that this boron-substituted diene can serve as a synthon for a host of organic dienes via cross coupling reactions which we performed on Diels–Alder reaction cycloadducts.
Experimental
Preparation of 1,3-butadiene-2-diethanolamine boronate 2: A mixture of magnesium (1.0 g, 41.1 mmol), 1,2-dibromoethane (0.5 mL), and THF (10 mL) was refluxed under nitrogen for 15 min to activate the magnesium. To the mixture anhydrous zinc chloride (0.6 g) in THF (60 mL) was added and reflux was continued for another 15 min. 2-Chloro-1,3-butadiene (4.9 mL, 25 mmol) (density 0.915 g/mL, 50% in xylene) and 1,2-dibromoethane (0.95 g, 5 mmol) in THF (30 mL) were added dropwise over a period of 30 min. This addition was controlled so as to bring the mixture into a gentle reflux. The color of the contents changed gradually from grayish white to greenish black. The mixture was heated to reflux for an additional 30 min after completion of the addition. The Grignard reagent thus obtained was immediately added dropwise to a solution of trimethoxyborane (4.25 mL, 38.5 mmol) in THF (25 mL) using a double-ended needle. The addition was controlled in such a way that the internal temperature of the mixture was maintained below –60 °C all the time. After completion of the addition, the solution was allowed to warm to room temperature quickly. The cloudy gray colored reaction mixture was stirred for 1 h. To the resulting mixture at room temperature, 0.5 M HCl solution (100 mL) was added. The reaction mixture was extracted with Et2O (2 × 75 mL). The combined colorless clear organic layers were dried over MgSO4, and the volatiles were removed by a rotary evaporator (30 °C, 20 Torr) to yield the dieneboronic acid. The boronic acid was added at once to a solution of diethanolamine (0.8 equiv, 22.5 mmol, 8.411g) dissolved in THF (100 mL). Sodium sulfate (8 g) was added and refluxed for 6 h. At the end of the reaction, the flask was cooled to room temperature. Solid Na2SO4 was separated from the solution by filtration. The solution was reduced by 50 mL using a rotary evaporator. A cold bath of −30 °C was used to induce crystallization. After 4 h, the solid was filtered and washed with cold chloroform. The product 2 was obtained as white needles (2.40 g, 14.4 mmol, 62.4%). 1H NMR (300 MHz, CDCl3) δ 6.51 (dd, J = 17.9, 10.9 Hz, 1H-H3), 5.46–5.40 (m, 3H), 4.98 (dd, J = 17.9, 1.9 Hz, 1H-H4), 5.18 (s, 1H-H7), 4.05 (m, 2H-H5,8), 3.89 (m, 2H-H5,8), 3.31 (m, 2H-H6,9), 2.76 (m, 2H-H6,9) 13C NMR (300, MHz, CDCl3) δ 143.6-C3, 124.3-C4, 114.6-C1, 63.4-C5,8, 52.1-C6,9, the signal of carbon C2 next to a tetravalent boron is generally not observed due to quadrupolar broadening [12]. Elemental anal. calcd for C8H14BNO2: C, 57.53; H, 8.45. Found: 57.06, 8.44.
Representative Diels–Alder procedure
Preparation of Diels–Alder product 3: Diene 2 (0.167 g, 1 mmol) and ethyl acrylate (0.700 g, 7 mmol) were dissolved in chloroform (15 mL) in a round bottomed flask and refluxed for 6 h. The white product was precipitated with pentane (150 mL) and obtained by vacuum filtration, (0.224 g, 0.84 mmol, 84%). 3: 1H NMR (300 MHz, CDCl3) δ 5.91 (m, 1H), 4.86 (s, 1H), 4.12 (q, J = 7.25, 2H), 3.97 (m, 2H), 2.893 (m, 2H), 3.224 (m, 2H), 2.79 (m, 2H), 2.48 (m, 1H), 2.23 (m, 2H), 2.11(m, 2H), 1.99 (m, 1H), 1.76 (s, 1H), 1.25 (t, J = 7.25, 3H). 13C NMR (300 MHz, CDCl3) δ Major isomer: 176.7, 139.9 (=C-B), 126.9, 62.85, 62.81, 60.0, 51.2, 40.1, 39.8, 28.6, 26.4, 25.9, 14.0. Minor isomer selected resonances: 176.2, 127.6, 24.7, 24.6. Major isomer: minor isomer = 16.4:1. Elemental anal. calcd. for C13H22BNO4: C, 58.45; H, 8.30. Found: 58.17, 8.32.
Representative Suzuki coupling procedure
General procedure: Boron compounds and iodoaromatic compounds were added to a N2 flushed flask with Pd2(dba)3 and K2CO3 in acetonitrile and ethanol (30 mL). The mixture was refluxed for 36 h and cooled to room temperature. The solution was filtered through silica gel to remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O (4 × 50 mL). The combined organic layers were dried over MgSO4 and volatiles were removed by rotary evaporation. The resulting cross-coupled cycloadduct residue was purified by flash chromatography (ethyl ether:hexane = 1:1). Optimization of conditions: 2% Pd2(dba)3 [Tris(dibenzylideneacetone)dipalladium (0)], acetonitrile:ethanol = 5:1, boron cycloadduct:iodoaromatic compounds = 1:2, K2CO3 (3 equiv) reaction time: 36 h.
Preparation of 6-(4-methoxyphenyl)-3a-methyl-2-phenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (14): Following the general procedure, 4-iodoanisole (0.234 g, 1 mmol) and 5 (0.178 g, 0.5 mmol) were added along with Pd2(dba)3 (10 mg) and K2CO3 (0.207 g, 1.5 mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and refluxed for 36 h. The resulting brown oily crude product mixture was subjected to flash chromatography to yield the cross-coupled product as a white solid (0.134 g, 0.39 mmol, 78%). 14: 1H NMR (300 MHz, CDCl3) δ Major isomer: 7.38 (d, J = 7.5 Hz, 2H), 7.31 (d, J = 8.7 Hz, 2H), 7.26 (m, 1H), 7.13 (d, J = 7.5 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 6.1 (m, 1H), 3.80 (s, 3H), 3.25 (dd, J = 15.4, 2.4 Hz, 1H), 2.99 (dd, J = 6.5, 2.4 Hz, 1H), 2.86 (dd, J = 15.4, 6.5 Hz, 1H), 2.61 (ddt, J = 15.4, 6.5, 2.4 Hz, 1H), 2.16 (dd, J = 15.4, 2.4 Hz, 1H), 1.50 (s, 3H). Minor isomer selected resonances: 3.15 (d, J = 15.4), 2.44 (m), 2.30 (m). 13C NMR (300 MHz, CDCl3) δ 182.3, 178.5, 159.5, 139.8, 133.1, 132.4, 129.4, 128.8, 127.0, 126.8, 122.0, 114.3, 55.6, 48.4, 45.1, 36.7, 30.6, 25.9. Elemental anal. calcd for C22H21NO3: C, 76.06; H, 6.09. Found: 76.34, 6.31.
Acknowledgements
We thank the National Science Foundation for their support of this work (CHE-0450722 and CHE-0749759) and the NMR instrumentation used to characterize the compounds reported here. The UNC Center for Mass Spectrometry performed high resolution mass spectral analyses. We thank Fred Salsbury of the Department of Physics for performing the DFT calculations discussed in this manuscript.
References
-
Smalley, T. L.; Wright, M. W.; Garmon, S. A.; Welker, M. E.; Rheingold, A. L. Organometallics 1993, 12, 998–1000. doi:10.1021/om00028a006
Return to citation in text: [1] -
Tada, M.; Shimizu, T. Bull. Chem. Soc. Jpn. 1992, 65, 1252–1256. doi:10.1246/bcsj.65.1252
Return to citation in text: [1] -
Pickin, K. A.; Kindy, J. M.; Day, C. S.; Welker, M. E. J. Organomet. Chem. 2003, 681, 120–133. doi:10.1016/S0022-328X(03)00587-4
Return to citation in text: [1] -
Welker, M. E. Curr. Org. Chem. 2001, 5, 785–807. doi:10.2174/1385272013375175
Return to citation in text: [1] -
Tucker, C. J.; Welker, M. E.; Day, C. S.; Wright, M. W. Organometallics 2004, 23, 2257–2262. doi:10.1021/om040010z
Return to citation in text: [1] -
Miyaki, Y.; Onishi, T.; Ogoshi, S.; Kurosawa, H. J. Organomet. Chem. 2000, 616, 135–139. doi:10.1016/S0022-328X(00)00583-0
Return to citation in text: [1] -
Chai, C. L. L.; Johnson, R. C.; Koh, J. Tetrahedron 2002, 58, 975–982. doi:10.1016/S0040-4020(01)01160-7
Return to citation in text: [1] -
De, S.; Welker, M. E. Org. Lett. 2005, 7, 2481–2484. doi:10.1021/ol050794s
Return to citation in text: [1] [2] -
De, S.; Day, C.; Welker, M. E. Tetrahedron 2007, 63, 10939–10948. doi:10.1016/j.tet.2007.08.063
Return to citation in text: [1] [2] -
Wright, M. W.; Smalley, T. L.; Welker, M. E.; Rheingold, A. L. J. Am. Chem. Soc. 1994, 116, 6777–6791. doi:10.1021/ja00094a037
Return to citation in text: [1] -
Pidaparthi, R. R.; Welker, M. E.; Day, C. S.; Wright, M. W. Org. Lett. 2007, 9, 1623–1626. doi:10.1021/ol070089e
Return to citation in text: [1] -
Darses, S.; Guillaume, M.; Genet, J.-P. Eur. J. Org. Chem. 1999, 8, 1875–1883. doi:10.1002/(SICI)1099-0690(199908)1999:8<1875::AID-EJOC1875>3.3.CO;2-N
Return to citation in text: [1]
| 1. | Smalley, T. L.; Wright, M. W.; Garmon, S. A.; Welker, M. E.; Rheingold, A. L. Organometallics 1993, 12, 998–1000. doi:10.1021/om00028a006 |
| 7. | Chai, C. L. L.; Johnson, R. C.; Koh, J. Tetrahedron 2002, 58, 975–982. doi:10.1016/S0040-4020(01)01160-7 |
| 6. | Miyaki, Y.; Onishi, T.; Ogoshi, S.; Kurosawa, H. J. Organomet. Chem. 2000, 616, 135–139. doi:10.1016/S0022-328X(00)00583-0 |
| 3. | Pickin, K. A.; Kindy, J. M.; Day, C. S.; Welker, M. E. J. Organomet. Chem. 2003, 681, 120–133. doi:10.1016/S0022-328X(03)00587-4 |
| 4. | Welker, M. E. Curr. Org. Chem. 2001, 5, 785–807. doi:10.2174/1385272013375175 |
| 5. | Tucker, C. J.; Welker, M. E.; Day, C. S.; Wright, M. W. Organometallics 2004, 23, 2257–2262. doi:10.1021/om040010z |
| 2. | Tada, M.; Shimizu, T. Bull. Chem. Soc. Jpn. 1992, 65, 1252–1256. doi:10.1246/bcsj.65.1252 |
| 10. | Wright, M. W.; Smalley, T. L.; Welker, M. E.; Rheingold, A. L. J. Am. Chem. Soc. 1994, 116, 6777–6791. doi:10.1021/ja00094a037 |
| 12. | Darses, S.; Guillaume, M.; Genet, J.-P. Eur. J. Org. Chem. 1999, 8, 1875–1883. doi:10.1002/(SICI)1099-0690(199908)1999:8<1875::AID-EJOC1875>3.3.CO;2-N |
| 9. | De, S.; Day, C.; Welker, M. E. Tetrahedron 2007, 63, 10939–10948. doi:10.1016/j.tet.2007.08.063 |
| 8. | De, S.; Welker, M. E. Org. Lett. 2005, 7, 2481–2484. doi:10.1021/ol050794s |
| 9. | De, S.; Day, C.; Welker, M. E. Tetrahedron 2007, 63, 10939–10948. doi:10.1016/j.tet.2007.08.063 |
| 11. | Pidaparthi, R. R.; Welker, M. E.; Day, C. S.; Wright, M. W. Org. Lett. 2007, 9, 1623–1626. doi:10.1021/ol070089e |
© 2009 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)