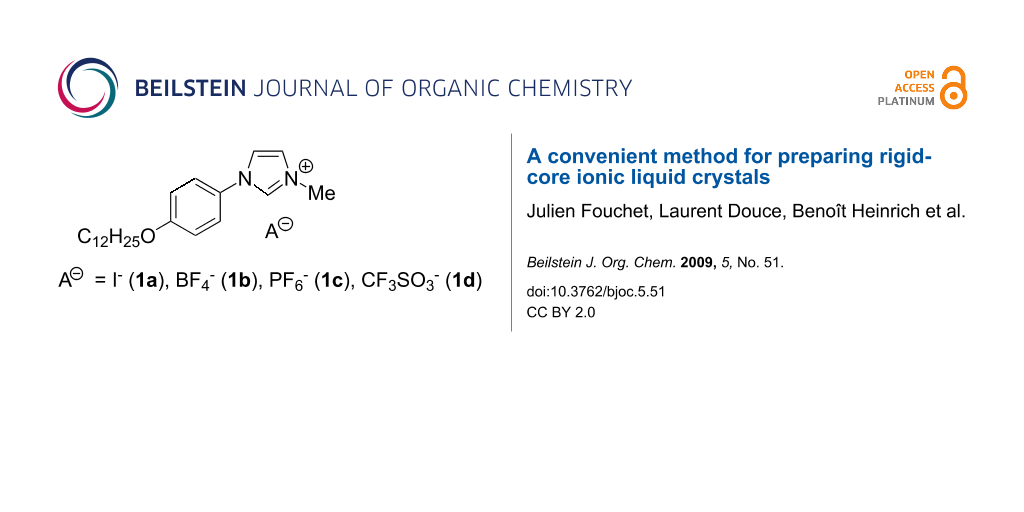Abstract
An efficient, solvent free method for the N-arylation of imidazole by 1-(dodecyloxy)-4-iodobenzene using Cu(II)-NaY as catalyst and K2CO3 as base is reported. By this synthetic approach, mesomorphic 3-[4-(dodecyloxy)phenyl]-1-methyl-1H-imidazol-3-ium iodide was synthesized in a two-step procedure, and its mesomorphism has been fully investigated by polarised optical microscopy, differential scanning calorimetry and X-ray diffraction. In addition its lamellar crystal structure, electrochemical behaviour and UV (absorption and emission) properties are reported.

Graphical Abstract
Introduction
Over the past decade extensive studies of ionic liquids (ILs) have revealed their many useful properties such as extremely low volatility, high thermal stability, non-flammability, high chemical and radiochemical stability, high ionic conductivity and wide electrochemical window [1-3]. In addition, the ILs have been used as reaction media increasing the yields of many syntheses and eliminating the hazards associated conventional solvents [4]. Thus are extremely versatile in that changes in both the cation and its counter anion can be used to finely tune their properties (for example: viscosity, melting point, polarity, hydrophilicity/hydrophobicity…). Important emerging applications include those in separation and extraction processes, and in various electrochemical devices, such as lithium ion batteries, fuel cells, and capacitors, as well as in synthesis and catalysis [1-5].
Liquid crystals are characterised by both mobility and self-organisation at the macroscopic level [6]. Almost all such mesomorphic materials are based on molecules combining two antagonistic units consisting of rigid (aromatic) and flexible (alkyl) or hydrophilic (polar heads) and hydrophobic (alkyl chains) parts. The subtle balance of their effects governs the formation of a multitude of supramolecular architectures depending on the temperature (thermotropic liquid crystals) and/or of the solvent (lyotropic liquid crystals) [7,8]. In the case of the thermotropic liquid crystals the arrangements give rise to nematic phases (molecules are aligned along an orientational axis), smectic phases (orientational/positional order in the layers) and columnar phases (orientational/positional order in the columns). The lyotropic compounds display not only lamellar and columnar organization but also hierarchical self-assembly in spheres (micelles), ribbons and fibres. These unique properties lead to their applications ranging from display technology through templating media for synthesis to biological activity (targeting and transporting of drugs and gene materials) [9].
Full convergence of the ionic liquid and liquid crystal fields could provide a vast range of materials (Ionic liquid crystals, ILCs) with novel and tunable characteristics such as those of ordered and oriented hybrid compound semiconductors exhibiting both electronic and ionic conductivity [10]. For this, the imidazolium unit is an excellent platform that can be designed to promote liquid crystalline phases and easily be doped by a large diversity of anions [11-21]. Variation of the N-substituents by Ullman coupling to extend the aromatic part is a facile means of creating this range [22,23].
Herein, we wish to report a solvent-free, N-arylation of imidazole as a means of expanding the aromatic core and obtaining unsymmetrical imidazolium liquid crystals (Scheme 1). We also describe the influence of the counter anion on the mesomorphism, electrochemistry and the UV properties of these imidazolium salts.
![[1860-5397-5-51-i1]](/bjoc/content/inline/1860-5397-5-51-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 1-[4-(dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium mesogenic salts.
Scheme 1: 1-[4-(dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium mesogenic salts.
Results and Discussion
Synthesis and characterization
Compound 1a was obtained in a two-step procedure. The first step was a coupling reaction between 1-(dodecyloxy)-4-iodobenzene and imidazole using Cu(II)-NaY as catalyst in the presence of potassium carbonate as base [23]. The reaction took place without solvent at 180 °C in a sealed tube over 72 h to afford 1-[4-(dodecyloxy)phenyl]-1H-imidazole (A) in a good (<80%) and reproducible yield (Scheme 2). Swager has already published the synthesis of compound A under standard Ullman conditions (K2CO3, CuI, L-proline in DMSO, 16 h at 110 °C) [22].
![[1860-5397-5-51-i2]](/bjoc/content/inline/1860-5397-5-51-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the imidazole A. Reaction conditions: (i) aryl iodide (1.37 mmol), imidazole (1.69 mmol), K2CO3 (1.51 mmol), Cu(II)NaY (148 mg), 72 h at 180 °C in a sealed tube.
Scheme 2: Synthesis of the imidazole A. Reaction conditions: (i) aryl iodide (1.37 mmol), imidazole (1.69 mmo...
The aryl-imidazole A was purified by column chromatography (ethyl acetate as eluent) on silica and characterized spectroscopically. The second step involved alkylation of A by iodomethane to give salt 1a in 89% yield after purification (Scheme 3). Distinctive signals for the CH (1H-imidazolium) group appear in the 1H and 13C NMR spectra at 10.45 ppm and 134.97 ppm respectively.
![[1860-5397-5-51-i3]](/bjoc/content/inline/1860-5397-5-51-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of methyl imidazolium 1a. Reaction conditions: (i) MeI in sealed tube, 54 h at RT.
Scheme 3: Synthesis of methyl imidazolium 1a. Reaction conditions: (i) MeI in sealed tube, 54 h at RT.
Single crystals of 1a suitable for X-ray diffraction were obtained by slow diffusion of ether into a CH2Cl2 solution. The compound 1a crystallizes in the triclinic space group P1. A partly labelled ORTEP view showing non-classical hydrogen bonds and C-H..π interactions is given in Figure 1 (the interactions also being listed in Table 1). The alkyloxy chains are quite parallel, as is clear from the crystal packing given in Figure 2, with segregation between the rigid part (including iodine atoms) and the alkyloxy chains (≈20 Å, see Figure 2). The length of the molecule in the crystalline state is about 24 Å.
![[1860-5397-5-51-1]](/bjoc/content/figures/1860-5397-5-51-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP view of compound 1a with partial labelling. The closest molecules are represented (with lower opacity) when connected by CH-π and/or non classical H-bonds (black thin lines). The ellipsoids enclose 50% of the electronic density. Symmetry operators for equivalent positions: d = ±1+x, y, z; e = 1+x, 1+y, z.
Figure 1: ORTEP view of compound 1a with partial labelling. The closest molecules are represented (with lower...
![[1860-5397-5-51-2]](/bjoc/content/figures/1860-5397-5-51-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Packing diagram of compound 1a in projection in the (b,c) lattice plane. Large spheres represent the iodine atoms.
Figure 2: Packing diagram of compound 1a in projection in the (b,c) lattice plane. Large spheres represent th...
Table 1: Non-classical hydrogen bonds and CH..π interactionsa occurring in 1a. Cg is the phenyl ring (C5 to C10). Symmetry operators for equivalent positions: d = ±1+x, y, z; e = 1+x, 1+y, z.
| C-H..I | dC-H (Å) | dH-I (Å) | dC-I (Å) | C-H-I (°) |
|---|---|---|---|---|
| C1-H1..Id | 0.95 | 2.8270 | 3.746(5) | 163.1 |
| C2-H2..Ie | 0.95 | 2.9123 | 3.822(5) | 160.6 |
| C11-H11B..I | 0.95 | 3.0026 | 3.992(5) | 179.2 |
| C-H..Cg | dC-H (Å) | dH-Cg (Å) | dC-I (Å) | C-H-Cg (°) |
| C4-H4A..Cgd | 0.95 | 3.109 | 3.502 | 105.6 |
| C4-H4B..Cgd | 0.95 | 3.309 | 3.502 | 93.1 |
| C17-H17B..Cge | 0.95 | 3.310 | 4.207 | 151.5 |
aPlaton software [24].
It should be emphasised that the lattice area (A = a·b·sin(γ) = 2V/d001 = 57.1 Å2) is about three times the transverse area of an all-trans crystallised chain and that even so the alkyl tails organise in segregated double layers, without interdigitation but with a tilt angle of 71° with respect to the layer normal. This large tilt angle just compensates the discrepancy between areas, maintaining the compactness of the packing and the flatness of the segregated ionic and aliphatic double layers. Apart from the crystallised state of the tails, this structure is very close to a smectic type of organisation. The segregation between the alkyl tails and the charged rigid parts indicates that by melting the chains they could show liquid crystal behaviour at a higher temperature. In order to understand the influence of the anion on the electrochemical, UV properties and mesomorphism, we prepared compounds with BF4− (1b), PF6− (1c), CF3SO3− (1d) and (CF3SO2)2N− (1e) anions in excellent yield by anion metathesis in water/CH2Cl2 as solvent (Scheme 4).
![[1860-5397-5-51-i4]](/bjoc/content/inline/1860-5397-5-51-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Anion metathesis in water/CH2Cl2 as solvent.
Scheme 4: Anion metathesis in water/CH2Cl2 as solvent.
All these compounds were fully characterized by 1H NMR, 13C NMR {1H}, FT-IR and UV spectroscopy, as well as elemental analysis. The IR spectra showed typical anion vibrations at 1024 cm−1 (1b BF4−), 826 cm−1 (1c PF6−), 1269 and 1028 cm−1 (1d CF3SO3−), 1358 and 1183 cm−1 (1e (CF3SO2)2N−). 1H NMR spectra were recorded in CDCl3, in which the chemical shift for the CH (1H-imidazolium) is very dependent upon the anion, with δ 10.45 (1a) 9.37 (1b), 9.10 (1c), 9.41 (1d) and 8.98 ppm (1e). This dependency is certainly due to the interactions though H-bonding and the charge localisation on the anion. The UV spectra displays typical charge transfer (π–π* or n–n*) transitions in CH2Cl2 at 240 nm (1a ε = 24000 M−1 cm−1), 255 nm (1b ε = 10500 M−1 cm−1), 249 nm (1c ε = 11700 M−1 cm−1), 256 nm (1d ε = 10100 M−1 cm−1) and 255 nm (1e ε = 11100 M−1 cm−1). A blue emission was also observed at 384 nm (Figure 3).
![[1860-5397-5-51-3]](/bjoc/content/figures/1860-5397-5-51-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Spectra of absorption (red line) and emission (blue line) of 1a.
Figure 3: Spectra of absorption (red line) and emission (blue line) of 1a.
Investigation of the Liquid Crystalline Behaviour
The thermogravimetric analysis of compounds 1a–e showed the general stability order to be I−< BF4− ≈ PF6−< CF3SO3−< (CF3SO2)2N− (Figure 4).
![[1860-5397-5-51-4]](/bjoc/content/figures/1860-5397-5-51-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: TGA measurements of the compounds 1a–e (rate 10 °C·min−1, in air).
Figure 4: TGA measurements of the compounds 1a–e (rate 10 °C·min−1, in air).
For all the compounds, the mesomorphic behaviour and phase transition temperatures were investigated by polarized optical microscopy (POM), differential scanning calorimetry (DSC), and powder X-ray diffractometry (XRD). To avoid possible effects of hydration of the materials, all were dried in vacuo before X-ray and DSC analyses. The phase transition temperatures and the corresponding enthalpy changes derived for compounds 1a–e are compiled in Table 2, while typical results are displayed in Figure 5.
Table 2: Phase transition temperatures and corresponding enthalpies determined from the 2nd heating and cooling.
| Anions | Phase |
Temperature
Cr ![[Graphic 1]](/bjoc/content/inline/1860-5397-5-51-i5.svg?max-width=637&scale=1.18182) LC LC
|
Phase |
Temperature
LC I
|
Phase |
|---|---|---|---|---|---|
| I− | Crystal |
113 °C (15.83 kJ/mol)
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-51-i6.svg?max-width=637&scale=1.0)
81 °C (12.76 kJ/mol) |
Smectic A |
250 °C
![[Graphic 3]](/bjoc/content/inline/1860-5397-5-51-i7.svg?max-width=637&scale=1.0)
|
Decomposition |
| BF4− | Crystal |
91 °C (30.08 kJ/mol)
60 °C (10.62 kJ/mol) |
Smectic A |
230 °C
|
Decomposition |
| PF6− | Crystal |
97 °C (36.21 kJ/mol)
68 °C (43.34 kJ/mol) |
Smectic A |
163 °C (1.12 kJ/mol)
163 °C (1.76 kJ/mol) |
Liquid |
| F3CO3− | Crystal |
77 °C (43.28 kJ/mol)
49 °C (44.00 kJ/mol) |
Smectic A |
95 °C (0.79 kJ/mol)
95 °C (1.04 kJ/mol) |
Liquid |
| (F3CSO2)2N− | Crystal |
59 °C (61.17 kJ/mol)
39 °C (61.12 kJ/mol) |
Liquid | ||
Legend: Cr: Crystal, LC: Liquid Crystal, I: Isotropic Liquid.
The high stability of the compounds was also demonstrated by the absence of significant perturbation of the DSC patterns following several heating–cooling cycles. Compounds 1e, not unexpectedly, do not show thermotropic behaviour, while the data for 1a–d give an order of anion stabilisation of liquid crystal behaviour of Br− > BF4− > PF6− > CF3SO3− (see Figure 5).
![[1860-5397-5-51-5]](/bjoc/content/figures/1860-5397-5-51-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Phase transition temperatures of compounds 1a–e.
Figure 5: Phase transition temperatures of compounds 1a–e.
The optical textures observed during slow cooling from isotropic melt showed the emergence of a smectic A phase (appearance of Batônnet rods, turning into a wide, fan-like, focal-conic texture). The smectic structure of the liquid crystal phase was confirmed by XRD. The X-ray pattern (Figure 6) of the Smectic A form recorded at 120 °C contains a diffuse band at 4.6 Å (wide angle), which shows clearly that the alkyl chains have a liquid-like structure and are segregated from the aromatic cores.
![[1860-5397-5-51-6]](/bjoc/content/figures/1860-5397-5-51-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Powder X-ray diffraction pattern of compound 1a in the liquid crystal state (T = 120 °C).
Figure 6: Powder X-ray diffraction pattern of compound 1a in the liquid crystal state (T = 120 °C).
The layer thickness in the Smectic A phase was determined from the position of the sharp reflection in the small angle region (d = 39.8 Å at 120 °C) and corresponds to the alternation between the sublayer formed by the molten chains and the sublayer formed by the ionic double layer and the mesogenic parts. The thickness of the corresponding sublayers’ alternation in the crystalline phase is given by the location of the d001 reflection (d001 = 20.21 Å from single crystal pattern at room temperature). Despite the enormous difference in layer thicknesses between both phases (the extrapolation of the variation versus temperature gives d = 46.5 Å at 20 °C i.e. more than twice d001), the difference in molecular volume (smectic phase: Vmol = 622 Å3 at 20 °C; crystalline phase: Vmol = V/2 = 577 Å3) just coincides with the contribution of the chain melting [25,26], indicating that the partial volume of the ionic sublayer does not change significantly between both phases. The observed layer thickness change is therefore the consequence of the different “molecular areas” S, i.e. the projection area of a mesogen counter-ion assembly within the mean smectic plane (S = 2Vmol/d), which is identical to the lattice area in the crystalline phase (S = V/d001). Thus, since no significant volume change is involved in the shrinking of S from 57.1 Å2 in the crystalline phase to 27 Å2 in the smectic A phase (value at 20 °C obtained from the extrapolation of the variation of S versus temperature), the ionic sublayer thickness dc (determined as dc = 2[Vmol/−Vch]/S, Vch being the chain volume) simultaneously expands in proportion (from 9.5 Å in the crystalline phase to 20 Å in the smectic phase at 20 °C). These lateral shrinkage and longitudinal extension events are the result of a ruffling process of the ionic sublayers, starting from the completely flat state in the crystalline phase shown by the single crystal structure (see Figure 7).
![[1860-5397-5-51-7]](/bjoc/content/figures/1860-5397-5-51-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The melting process involves the ruffling of the ionic sublayer. In the smectic phase, the ruffling degree decreases with increasing temperature.
Figure 7: The melting process involves the ruffling of the ionic sublayer. In the smectic phase, the ruffling...
The maximum degree of ruffling in the smectic A phase is reached just before crystallisation, since the experimental temperature dependence of S and dc indicates that the sublayers continuously spread with increasing temperature (see Figure 8).
![[1860-5397-5-51-8]](/bjoc/content/figures/1860-5397-5-51-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Comparison of the molecular area S and of the ionic sublayer thickness dc (including mesogenic segments) in the crystalline phase (circle) and in the smectic A phase (squares) for compound 1a (I−).
Figure 8: Comparison of the molecular area S and of the ionic sublayer thickness dc (including mesogenic segm...
The counter-ion substitution within the series 1 involves large changes of S, but the temperature dependence and dc values are roughly the same for all terms (see Figure 9).
![[1860-5397-5-51-9]](/bjoc/content/figures/1860-5397-5-51-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Variation with the counter-ion of the molecular area S and of the ionic sublayer thickness dc (including mesogenic segments) in the smectic A phase for series 1: squares: 1a (I−); circles: 1b (BF4−); up triangles: 1c (PF6−); down triangles: 1d (CF3SO3−).
Figure 9: Variation with the counter-ion of the molecular area S and of the ionic sublayer thickness dc (incl...
The influence of the substitution can therefore be considered as an anion size effect, since the lattice area expands with increasing counter-ion bulkiness without change of the degree of ruffling (the small discrepancies for compound 1d (CF3SO3−) being explained by the presence of the CF3 lateral group, which contributes to dc and perturbates slightly the interface with the aliphatic sublayer). It should be emphasised that the stability of the smectic A phase is not determined by the degree of ruffling of the ionic sublayer but by the folding degree of the tails and therefore the thickness of the aliphatic sublayers. Thus, depending upon the anion size, the isotropisation occurs at various temperatures, but for approximately the same maximum molecular area (Smax ≈ 41 Å2) and therefore similar minimum aliphatic sublayer thicknesses (dchmin ≈ 19 Å).
To summarise, the large discrepancy between the lattice area and the cross section of the aliphatic chains are taken into account differently in the crystalline and in the smectic molecular organisations. In the crystalline phase, the ionic sublayers just impose their area and the tails tilt until dense packing is reached. In the smectic phase, tail tilting is not favourable upon the amphipathic expelling at the interface with the ionic sublayer and the system adopts a compromise molecular area associating ruffled ionic sublayers and folded aliphatic tails. With increasing temperature, the aliphatic chains spread more easily and the organisation shifts toward flat sublayers. A more detailed investigation of the molecular area variation in series involving both, counter-ion substitution and tail-length variation, has been presented elsewhere for a very similar cationic structure [20,25,26].
Electrochemical behaviour
Cyclic voltammetry was used to determine the electrochemical behaviour of the compounds 1a, 1b and 1c, the voltammograms being recorded in CH3CN solutions containing 0.1 M NBu4PF6 as supporting electrolyte at a platinum working electrode. The peak potentials are given vs. a SCE. Representative cyclic voltammograms of 1a are shown in Figure 10. The anodic portion of the voltage scan displays two oxidation steps having peak potentials of 0.42 V and 0.68 V, and likely involve the formation of I2 and possibly then a higher-oxidation-state iodine (I3−) species. As seen for 1a (Figure 9), for 1b and 1c the cathodic portion of the voltage scan displays only an irreversible reduction step at ca 1.58 V, which corresponds to the reduction of the cationic imidazolium species. Note that the peak at −0.8 V is probably due to the reduction of O2 which is difficult to eliminate from the solution.
![[1860-5397-5-51-10]](/bjoc/content/figures/1860-5397-5-51-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Cyclic voltammogram of 1a in CH3CN (0.1 M NBu4PF6): (i), (ii) cathodic and anodic range of the voltage scan. Scan rate 100 mV·s−1. The black star denotes the initial and final potential.
Figure 10: Cyclic voltammogram of 1a in CH3CN (0.1 M NBu4PF6): (i), (ii) cathodic and anodic range of the volt...
Conclusion
In conclusion, we report synthetic methodology based on Ullman coupling to extend the imidazolium aromatic core. From this coupling product we have synthesized and fully characterized new mesomorphic compounds with different anions. We have also determined a structure by X-ray diffraction on a single crystal. The crystallisation shows the completely lamellar segregation between the flexible chains and the rigid part. The layers are linked to each other by the semi-interdigitation of alkyl tails. Despite an enormous difference between the cross-section of crystallised chains and the lattice area imposed by the organisation within the ionic sublayers, the latter just remain flat and the tails undergo a double layer dense packing with 71° tilting with respect to the layer normal. In the smectic phase, area matching is achieved by ruffling of the sublayers and folding of the molten aliphatic tails, the degree of ruffling decreasing with increasing temperature. The electrochemical windows have been measured and we are attempting to measure the carrier mobility in order to fully assess the prospects for using these molecules in molecular electronics. We intend also to introduce different length tails in order to obtain room temperature ionic liquid crystals, aswell as to explore use of the coupling reaction between imidazole and other aromatics and heterocycles to tune the electronic properties.
Experimental
X-ray diffraction pattern of powder samples in Lindeman capillaries or sealed cells were measured in transmission by using a focused CuKα1 linear beam, temperature control being within 0.03 °C and acquisition being conducted with an Inel CS120 curved counter. The molecular volumes of all compounds were calculated with an accuracy of 0.5% from the measurements performed for an analogous compound [26] and from the methylene and counter ion partial volumes.
All reagents were purchased from commercial suppliers and used without further purification. Chromatography was carried out with Merck silica gel 60 (40–63 mm). Analytical TLC was performed with Merck silica gel 60 F254 aluminium sheets. 1H NMR and 13C NMR (300 MHz and 75 MHz respectively) spectra were recorded with a Bruker Avance 300 spectrometer at 25 °C. Chemical shifts, δ, are reported in ppm using TMS as internal standard, spin-spin coupling constants, J, are given in Hz and the abbreviations s, br, s, t, q, m were used to denote respectively the multiplicity of signals: singlet, broad singlet, triplet, quadruplet, multiplet. Infrared spectra were recorded (KBr pastille) with a spectrophotometer IR Digital FTS 3000. UV/Vis spectra were recorded with a spectrophotometer U-3000. Elemental analyses were performed by the analytical service at the Institut Charles Sadron and by the analytical service at the Université de Strasbourg (Strasbourg, France). The optical structures of mesophases were studied with a Leitz polarizing microscope equipped with a Mettler FP80 hot stage and an FP80 central processor. The TGA measurements were carried out on a SDTQ 600 apparatus at scanning rate of 10 °C·min−1. The transition temperatures and enthalpies were measured by differential scanning calorimetry with a DSC Q1000 from TA Instruments at different temperature rates (5 °C·min−1, 2 °C·min−1) on heating and cooling.
1-[4-(Dodecyloxy)phenyl]-1H-imidazole (A)
1-Dodecyloxy-4-iodobenzene (0.533 g, 1.37 mmol), imidazole (0.115 g, 1.69 mmol), K2CO3 (0.288 g, 1.51 mmol) and Cu(II)-NaY (0.148 g) were heated in sealed tube to 180 °C for 72 h. The reaction mixture was filtered to remove the catalyst and the filtrate was purified by column chromatography (silica gel, ethyl acetate) to afford pure A (0.378 g, 84%).
1H NMR (300 MHz, CDCl3): δ = 0.88 (t, 3H, J = 6.5 Hz, CH3 aliphatic chain), 1.27 (broad s, 16H, CH2 aliphatic chain), 1.42–1.49 (m, 2H, O-CH2-CH2-CH2), 1.75–1.84 (m, 2H, O-CH2-CH2), 3.98 (t, 2H, J = 6.6 Hz, O-CH2), 6.96 and 7.27 (AA′ and BB′, 2 × 2H, J = 9.0 Hz, CH phenyl), 7.17–7.19 (m, 2H, N-CH-CH-N), 7.75 (broad s, 1H, N-CH-N). 13C NMR (75 MHz, CDCl3): δ = 14.03 (CH3 aliphatic), 22.61, 25.94, 29.13, 29.27, 29.30, 29.50, 29.52, 29.57, 29.59, 31.84 (CH2 aliphatic), 68.40 (O-CH2), 115. 39 (CH phenyl), 118.67 (CH imidazolium), 123.09 (CH phenyl), 129.97 (CH imidazolium), 130.47 (N-C phenyl), 135.79 (CH imidazolium), 158.48 (C-O-CH2 phenyl). νmax/cm−1 3118 (C-H aromatic), 2921 and 2851 (C-H aliphatic), 1520 (C=C aromatic), 1243 (C-O aromatic). UV/Vis (CH2Cl2): λmax (ε, L·mol−1·cm−1) = 241 nm (15000). Elemental analysis for C21H32N2O, Cacld: C, 76.78; H, 9.82; N, 8.53%. Found: C, 76.96; H, 10.58; N, 8.57%.
1-[4-(Dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium iodide (1a)
A mixture of A (1.069 g, 3.25 mmol) and iodomethane (2 mL, 31.80 mmol) was stirred in a sealed tube for 54 h and was heated to 40 °C for 10 minutes. Diethyl ether was added and the reaction mixture was filtered and the solid was washed with diethyl ether. Crystallization with dichloromethane and diethyl ether gave de 1a (1.318 g, 89%).
1H NMR (300 MHz, CDCl3): δ = 0.89 (t, 3H, J = 6.9 Hz, CH3 aliphatic chain), 1.28 (broad s, 16H, CH2 aliphatic chain), 1.41–1.48 (m, 2H, O-CH2-CH2-CH2), 1.76–1.85 (m, 2H, O-CH2-CH2), 3.99 (t, 2H, J = 6.6 Hz, O-CH2), 4.27 (s, 3H, N-CH3), 7.04 and 7.66 (AA′ and BB′, 2 × 2H, J = 9.1 Hz, CH phenyl), 7.46–7.48 (m, 2H, N-CH-CH-N), 10.45 (broad s, 1H, N-CH-N). 13C NMR (75 MHz, CDCl3): δ = 14.01 (CH3 aliphatic), 22.58, 25.89, 29.00, 29.25, 29.29, 29.48, 29.51, 29.54, 29.57, 31.82 (CH2 aliphatic), 37.57 (N-CH3), 68.65 (O-CH2), 115. 99 (CH phenyl), 121.03 (CH imidazolium), 123.65 (CH phenyl), 124.42 (CH imidazolium), 127.05 (N-C phenyl), 135.49 (CH imidazolium), 160.47 (C-O-CH2 phenyl). νmax/cm−1 3131 (C-H aromatic), 2921 and 2851 (C-H aliphatic), 1514 (C=C aromatic), 1251 (C-O aromatic). UV–vis (CH2Cl2): λmax (ε, L·mol−1·cm−1) = 240 nm (24000). Elemental analysis for C22H35IN2O·1/4H2O, Calcd: C, 55.64; H, 7.53; N, 5.90%. Found: C, 55.78; H, 7.48; N, 5.34%.
General procedure for metathesis in water–anion exchange
A mixture of 1a dissolved in dichloromethane (4 mL) and a mixture of the corresponding salts dissolved in water (3 mL) were stirred together for 140 h. The organic layer was separated off, washed with water and dried over calcium chloride. Crystallization with dichloromethane and diethyl ether gave the corresponding imidazolium salt.
1-[4-(Dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium tetrafluoroborate (1b)
Following the general procedure using 1a (0.797 g, 1.69 mmol) and sodium tetrafluoroborate (0.511 g, 4.56 mmol) provided 1b with a yield of 72% (0.525 g, 1.22 mmol).
1H NMR (300 MHz, CDCl3): δ = 0.86 (t, 3H, J = 6.3 Hz, CH3 aliphatic chain), 1.28 (broad s, 16H, CH2 aliphatic chain), 1.41–1.46 (m, 2H, O-CH2-CH2-CH2), 1.75–1.84 (m, 2H, O-CH2-CH2), 3.98 (t, 2H, J = 6.3 Hz, O-CH2), 4.11 (s, 3H, N-CH3), 7.02 and 7.53 (AA′ and BB′, 2 × 2H, J = 9.0 Hz, CH phenyl), 7.48 (broad s, 2H, N-CH-CH-N), 9.37 (broad s, 1H, N-CH-N). 13C NMR (75 MHz, CDCl3): δ = 14.07 (CH3 aliphatic), 22.65, 25.95, 29.08, 29.32, 29.37, 29.55, 29.59, 29.61, 29.64, 31.89 (CH2 aliphatic), 36.81 (N-CH3), 68.65 (O-CH2), 115. 96 (CH phenyl), 121.34 (CH imidazolium), 123.59 (CH phenyl), 124.50 (CH imidazolium), 127.24 (N-C phenyl), 134.97 (CH imidazolium), 160.47 (C-O-CH2 phenyl). νmax/cm−1 2917 and 2849 (C-H aliphatic), 1514 (C=C aromatic), 1249 (C-O aromatic), 1024 (BF4−). UV–vis (CH2Cl2): λmax (ε, L·mol−1·cm−1) = 255 nm (10500). Elemental analysis for C22H35BF4N2O·3/4H2O, Calcd: C 59.53, H 8.29, N 6.31%. Found: C 59.74, H 8.02, N 6.20%.
1-[4-(Dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium hexafluorophosphate (1c)
Following the general procedure using 1a (0.695 g, 1.48 mmol) and potassium hexafluoroborate (0.518 g, 2.18 mmol) provided 1c with a yield of 84% (0.607 g, 1.24 mmol).
1H NMR (300 MHz, CDCl3): δ = 0.89 (t, 3H, J = 6.8 Hz, CH 3 aliphatic chain), 1.28 (broad s, 16H, CH2 aliphatic chain), 1.41–1.46 (m, 2H, O-CH2-CH2-CH2), 1.76–1.85 (m, 2H, O-CH2-CH2), 3.98 (t, 2H, J = 6.6 Hz, O-CH2), 4.07 (s, 3H, N-CH3), 7.02 and 7.48 (AA′ and BB′, 2 × 2H, J = 8.8 Hz, CH phenyl), 7.45 (broad s, 2H, N-CH-CH-N), 9.10 (broad s, 1H, N-CH-N). 13C NMR (75 MHz, CDCl3): δ = 14.06 (CH3 aliphatic), 22.65, 25.95, 29.08, 29.32, 29.37, 29.55, 29.59, 29.61, 29.64, 31.89 (CH2 aliphatic), 36.81 (N-CH3), 68.65 (O-CH2), 115. 91 (CH phenyl), 121.60 (CH imidazolium), 123.70 (CH phenyl), 124.33 (CH imidazolium), 127.21 (N-C phenyl), 134.42 (CH imidazolium), 160.51 (C-O-CH2 phenyl). νmax/cm−1 2921 and 2850 (C-H aliphatic), 1516 (C=C aromatic), 1255 (C-O aromatic), 826 cm−1 (PF6−). UV–vis (CH2Cl2): λmax (ε, L·mol−1·cm−1) = 249 nm (11700). Elemental analysis for C22H35F6N2OP·1/7H2O, Calcd: C 53.81, H 7.24, N 5.70%. Found: C 53.77, H 7.31, N 5.51%.
1-[4-(Dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium trifluoromethanesulfonate (1d)
Following the general procedure using 1a (0.730 g, 1.55 mmol) and sodium trifluoromethanesulfonate (0.616 g, 3.51 mmol) provided 1d with a yield of 46% (0.349 g, 0.71 mmol).
1H NMR (300 MHz, CDCl3): δ = 0.89 (t, 3H, J = 6.8Hz, CH3 aliphatic chain), 1.28 (broad s, 16H, CH2 aliphatic chain), 1.41–1.46 (m, 2H, O-CH2-CH2-CH2), 1.76–1.83 (m, 2H, O-CH2-CH2), 3.99 (t, 2H, J = 6.6 Hz, O-CH2), 4.10 (s, 3H, N-CH3), 7.02 and 7.51 (AA′ and BB′, 2 × 2H, J = 8.8 Hz, CH phenyl), 7.49 (broad s, 2H, N-CH-CH-N), 9.41 (broad s, 1H, N-CH-N). 13C NMR (75 MHz, CDCl3): δ = 13.96 (CH3 aliphatic), 22.54, 25.85, 28.97, 29.21, 29.26, 29.44, 29.48, 29.51, 29.53, 31.78 (CH2 aliphatique), 36.52 (N-CH3), 68.54 (O-CH2), 115. 84 (CH phenyl), 120.49 (q, J = 318.18 Hz, CF3SO3−), 121.36 (CH imidazolium), 123.33 (CH phenyl), 124.44 (CH imidazolium), 127.15 (N-C phenyl), 134.95 (CH imidazolium), 160.36 (C-O-CH2 phenyl). νmax/cm−1 3119 (C-H aromatic), 2915 and 2849 (C-H aliphatic), 1520 (C=C aromatic), 1269 and 1028 (CF3SO3−). UV–vis (CH2Cl2): λmax (ε, L·mol−1·cm−1) = 256 nm (10100). Elemental analysis for C22H35F3N2O4S, Cacld: C 56.08, H 7.16, N 6.59%. Found: C 55.84, H 6.86, N 5.40%.
1-[4-(Dodecyloxy)phenyl]-3-methyl-1H-imidazol-3-ium bis(trifluoromethane) sulfonamide (1e)
1a (0.101 g, 0.21 mmol) and lithium bis(trifluoromethane)sulfonamide (0.145 g, 0.51 mmol) were dissolved in water (3 mL) and stirred for 140 h. The precipitate was filtred and washed. Crystallization (chloroform/cyclohexane) provided 1e with a yield of 90% (0.121 g, 0.19 mmol).
1H NMR (300 MHz, CDCl3): δ = 0.89 (t, 3H, J = 6.8 Hz, CH3 aliphatic chain), 1.28 (broad s, 16H, CH2 aliphatic chain), 1.42–1.52 (m, 2H, O-CH2-CH2-CH2), 1.77–1.86 (m, 2H, O-CH2-CH2), 4.01 (t, 2H, J = 6.6 Hz, O-CH2), 4.07 (s, 3H, N-CH3), 7.05 and 7.46 (AA′ and BB′, 2 × 2H, J = 8.8 Hz, CH phenyl), 7.43–7.49 (m, 2H, N-CH-CH-N), 8.98 (broad s, 1H, N-CH-N). 13C NMR (75 MHz, CDCl3): δ = 14.06 (CH3 aliphatic), 22.65, 25.94, 29.05, 29.31, 29.34, 29.53, 29.57, 29.60, 29.63, 31.89 (CH2 aliphatic), 36.69 (N-CH3), 68.70 (O-CH2), 116.04 (CH phenyl), 119.77 (q, J = 319.29 Hz, CF3SO3−), 121.89 (CH imidazolium), 123.77 (CH phenyl), 124.31 (CH imidazolium), 126.99 (N-C phenyl), 134.67 (CH imidazolium), 160.78 (C-O-CH2 phenyl). νmax/cm−1 2918 and 2850 (C-H aliphatic), 1517 (C=C aromatic), 1358 cm−1 and 1183 ((CF3SO2)2N−). UV–vis (CH2Cl2): λmax (ε, L·mol−1·cm−1) = 255 nm (11100). Elemental analysis for C24H35F6N3O5S2.1/2H2O, Cacld: C 45.56, H 5.74, N 6.64%. Found: C 45.52, H 5.66, N 6.58%.
References
-
Wasserscheid, P.; Welton, T. Ionic Liquid in Synthesis; Wiley-VCH: Weinheim, Germany, 2003.
Return to citation in text: [1] [2] -
Rogers, R. B.; Seddon, K. R., Eds. Ionic Liquids IIIA: Fundamentals, Progress, Challenges and Opportunities, American Chemical Society, Symp. Ser.: Washington DC, 2005. doi:10.1021/bk-2005-0901
Return to citation in text: [1] [2] -
Rogers, R. B.; Seddon, K. R., Eds. Ionic Liquids IIIB: Fundamentals, Progress, Challenges and Opportunities, American Chemical Society, Symp. Ser.: Washington DC, 2005. doi:10.1021/bk-2005-0902
Return to citation in text: [1] [2] -
Dupont, J.; de Souza, R. F.; Suarez, P. A. Chem. Rev. 2002, 102, 3667–3692. doi:10.1021/cr010338r
Return to citation in text: [1] [2] -
Endres, F.; Abbott, A. P.; MacFarlane, D. R. Electrodeposition from Ionic liquids; Wiley-VCH: Weinheim, 2008. doi:10.1002/9783527622917
Return to citation in text: [1] -
Handbook of Liquid Crystals, Vols. 1, 2a, 2b and 3. Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: Weinheim, 1998.
Return to citation in text: [1] -
Guillon, D. Struct. Bond. 1999, 95, 41–82. doi:10.1007/3-540-68118-3_2
Return to citation in text: [1] -
Goodby, J. W.; Saez, I. M.; Cowling, S. J.; Görtz, V.; Draper, M.; Hall, A. W.; Sia, S.; Cosquer, G.; Lee, S.-E.; Raynes, E. P. Angew. Chem., Int. Ed. 2008, 47, 2754–2787. doi:10.1002/anie.200701111
Return to citation in text: [1] -
Dobbs, W.; Heinrich, B.; Bourgogne, C.; Donnio, B.; Terazzi, E.; Bonnet, M.-E.; Stock, F.; Erbacher, P.; Bolcato-Bellemin, A.-L.; Douce, L. J. Am. Chem. Soc. 2009, 131, 13338–13346. doi:10.1021/ja903028f
Return to citation in text: [1] -
Ohno, H. Electrochemical Aspect of Ionic Liquids; John Wiley and Sons, 2005. doi:10.1002/0471762512
Return to citation in text: [1] -
Binnemans, K. Chem. Rev. 2005, 105, 4148–4204. doi:10.1021/cr0400919
Return to citation in text: [1] -
Bowlas, C. J.; Bruce, D. W.; Seddon, K. R. Chem. Commun. 1996, 1625–1626. doi:10.1039/cc9960001625
Return to citation in text: [1] -
Yoshio, M.; Mukai, T.; Ohno, H.; Kato, T. J. Am. Chem. Soc. 2004, 126, 994–995. doi:10.1021/ja0382516
Return to citation in text: [1] -
Taubert, A. Angew. Chem., Int. Ed. 2004, 43, 5380–5382. doi:10.1002/anie.200460846
Return to citation in text: [1] -
Suisse, J.-M.; Bellemin-Laponnaz, S.; Douce, L.; Maisse-François, A.; Welter, R. Tetrahedron Lett. 2005, 46, 4303–4305. doi:10.1016/j.tetlet.2005.04.114
Return to citation in text: [1] -
Dobbs, W.; Suisse, J.-M.; Douce, L.; Welter, R. Angew. Chem., Int. Ed. 2006, 45, 4179–4182. doi:10.1002/anie.200600929
Return to citation in text: [1] -
Dobbs, W.; Douce, L.; Allouche, L.; Louati, A.; Malbosc, F.; Welter, R. New J. Chem. 2006, 30, 528–532. doi:10.1039/b600279j
Return to citation in text: [1] -
Suisse, J.-M.; Douce, L.; Bellemin-Laponnaz, S.; Maisse-François, A.; Welter, R.; Miyake, Y.; Shimizu, Y. Eur. J. Inorg. Chem. 2007, 3899–3905. doi:10.1002/ejic.200700251
Return to citation in text: [1] -
Yazaki, S.; Kamikawa, Y.; Yoshio, M.; Hamasaki, A.; Mukai, T.; Ohno, H.; Kato, T. Chem. Lett. 2008, 37, 538–539. doi:10.1246/cl.2008.538
Return to citation in text: [1] -
Yoshio, M.; Ichikawa, T.; Shimura, H.; Kagata, T.; Hamasaki, A.; Mukai, T.; Ohno, H.; Kato, T. Bull. Chem. Soc. Jpn. 2007, 80, 1836–1841. doi:10.1246/bcsj.80.1836
Return to citation in text: [1] [2] -
Fanta, P. E. Synthesis 1974, 9–21. doi:10.1055/s-1974-23219
Return to citation in text: [1] -
Kouwer, P. H. J.; Swager, T. M. J. Am. Chem. Soc. 2007, 129, 14042–14052. doi:10.1021/ja075651a
Return to citation in text: [1] [2] -
Kantam, M. L.; Rao, B. P. C.; Choudary, B. M.; Reddy, R. S. Synlett 2006, 14, 2195–2198. doi:10.1055/s-2006-949615
Return to citation in text: [1] [2] -
Spek, A. L. J. Appl. Crystallogr. 2003, 36, 7–13. doi:10.1107/S0021889802022112
Return to citation in text: [1] -
Cruz, C.; Heinrich, B.; Ribeiro, A. C.; Bruce, D. W.; Guillon, D. Liq. Cryst. 2000, 27, 1625–1631. doi:10.1080/026782900750037185
Return to citation in text: [1] [2] -
Dobbs, W.; Heinrich, B.; Douce, L. Beilstein J. Org. Chem. 2009, 5, No. 62. doi:10.3762/bjoc.5.62
Return to citation in text: [1] [2] [3]
| 1. | Wasserscheid, P.; Welton, T. Ionic Liquid in Synthesis; Wiley-VCH: Weinheim, Germany, 2003. |
| 2. | Rogers, R. B.; Seddon, K. R., Eds. Ionic Liquids IIIA: Fundamentals, Progress, Challenges and Opportunities, American Chemical Society, Symp. Ser.: Washington DC, 2005. doi:10.1021/bk-2005-0901 |
| 3. | Rogers, R. B.; Seddon, K. R., Eds. Ionic Liquids IIIB: Fundamentals, Progress, Challenges and Opportunities, American Chemical Society, Symp. Ser.: Washington DC, 2005. doi:10.1021/bk-2005-0902 |
| 7. | Guillon, D. Struct. Bond. 1999, 95, 41–82. doi:10.1007/3-540-68118-3_2 |
| 8. | Goodby, J. W.; Saez, I. M.; Cowling, S. J.; Görtz, V.; Draper, M.; Hall, A. W.; Sia, S.; Cosquer, G.; Lee, S.-E.; Raynes, E. P. Angew. Chem., Int. Ed. 2008, 47, 2754–2787. doi:10.1002/anie.200701111 |
| 26. | Dobbs, W.; Heinrich, B.; Douce, L. Beilstein J. Org. Chem. 2009, 5, No. 62. doi:10.3762/bjoc.5.62 |
| 6. | Handbook of Liquid Crystals, Vols. 1, 2a, 2b and 3. Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: Weinheim, 1998. |
| 1. | Wasserscheid, P.; Welton, T. Ionic Liquid in Synthesis; Wiley-VCH: Weinheim, Germany, 2003. |
| 2. | Rogers, R. B.; Seddon, K. R., Eds. Ionic Liquids IIIA: Fundamentals, Progress, Challenges and Opportunities, American Chemical Society, Symp. Ser.: Washington DC, 2005. doi:10.1021/bk-2005-0901 |
| 3. | Rogers, R. B.; Seddon, K. R., Eds. Ionic Liquids IIIB: Fundamentals, Progress, Challenges and Opportunities, American Chemical Society, Symp. Ser.: Washington DC, 2005. doi:10.1021/bk-2005-0902 |
| 4. | Dupont, J.; de Souza, R. F.; Suarez, P. A. Chem. Rev. 2002, 102, 3667–3692. doi:10.1021/cr010338r |
| 5. | Endres, F.; Abbott, A. P.; MacFarlane, D. R. Electrodeposition from Ionic liquids; Wiley-VCH: Weinheim, 2008. doi:10.1002/9783527622917 |
| 25. | Cruz, C.; Heinrich, B.; Ribeiro, A. C.; Bruce, D. W.; Guillon, D. Liq. Cryst. 2000, 27, 1625–1631. doi:10.1080/026782900750037185 |
| 26. | Dobbs, W.; Heinrich, B.; Douce, L. Beilstein J. Org. Chem. 2009, 5, No. 62. doi:10.3762/bjoc.5.62 |
| 4. | Dupont, J.; de Souza, R. F.; Suarez, P. A. Chem. Rev. 2002, 102, 3667–3692. doi:10.1021/cr010338r |
| 20. | Yoshio, M.; Ichikawa, T.; Shimura, H.; Kagata, T.; Hamasaki, A.; Mukai, T.; Ohno, H.; Kato, T. Bull. Chem. Soc. Jpn. 2007, 80, 1836–1841. doi:10.1246/bcsj.80.1836 |
| 25. | Cruz, C.; Heinrich, B.; Ribeiro, A. C.; Bruce, D. W.; Guillon, D. Liq. Cryst. 2000, 27, 1625–1631. doi:10.1080/026782900750037185 |
| 26. | Dobbs, W.; Heinrich, B.; Douce, L. Beilstein J. Org. Chem. 2009, 5, No. 62. doi:10.3762/bjoc.5.62 |
| 22. | Kouwer, P. H. J.; Swager, T. M. J. Am. Chem. Soc. 2007, 129, 14042–14052. doi:10.1021/ja075651a |
| 23. | Kantam, M. L.; Rao, B. P. C.; Choudary, B. M.; Reddy, R. S. Synlett 2006, 14, 2195–2198. doi:10.1055/s-2006-949615 |
| 22. | Kouwer, P. H. J.; Swager, T. M. J. Am. Chem. Soc. 2007, 129, 14042–14052. doi:10.1021/ja075651a |
| 11. | Binnemans, K. Chem. Rev. 2005, 105, 4148–4204. doi:10.1021/cr0400919 |
| 12. | Bowlas, C. J.; Bruce, D. W.; Seddon, K. R. Chem. Commun. 1996, 1625–1626. doi:10.1039/cc9960001625 |
| 13. | Yoshio, M.; Mukai, T.; Ohno, H.; Kato, T. J. Am. Chem. Soc. 2004, 126, 994–995. doi:10.1021/ja0382516 |
| 14. | Taubert, A. Angew. Chem., Int. Ed. 2004, 43, 5380–5382. doi:10.1002/anie.200460846 |
| 15. | Suisse, J.-M.; Bellemin-Laponnaz, S.; Douce, L.; Maisse-François, A.; Welter, R. Tetrahedron Lett. 2005, 46, 4303–4305. doi:10.1016/j.tetlet.2005.04.114 |
| 16. | Dobbs, W.; Suisse, J.-M.; Douce, L.; Welter, R. Angew. Chem., Int. Ed. 2006, 45, 4179–4182. doi:10.1002/anie.200600929 |
| 17. | Dobbs, W.; Douce, L.; Allouche, L.; Louati, A.; Malbosc, F.; Welter, R. New J. Chem. 2006, 30, 528–532. doi:10.1039/b600279j |
| 18. | Suisse, J.-M.; Douce, L.; Bellemin-Laponnaz, S.; Maisse-François, A.; Welter, R.; Miyake, Y.; Shimizu, Y. Eur. J. Inorg. Chem. 2007, 3899–3905. doi:10.1002/ejic.200700251 |
| 19. | Yazaki, S.; Kamikawa, Y.; Yoshio, M.; Hamasaki, A.; Mukai, T.; Ohno, H.; Kato, T. Chem. Lett. 2008, 37, 538–539. doi:10.1246/cl.2008.538 |
| 20. | Yoshio, M.; Ichikawa, T.; Shimura, H.; Kagata, T.; Hamasaki, A.; Mukai, T.; Ohno, H.; Kato, T. Bull. Chem. Soc. Jpn. 2007, 80, 1836–1841. doi:10.1246/bcsj.80.1836 |
| 21. | Fanta, P. E. Synthesis 1974, 9–21. doi:10.1055/s-1974-23219 |
| 24. | Spek, A. L. J. Appl. Crystallogr. 2003, 36, 7–13. doi:10.1107/S0021889802022112 |
| 10. | Ohno, H. Electrochemical Aspect of Ionic Liquids; John Wiley and Sons, 2005. doi:10.1002/0471762512 |
| 9. | Dobbs, W.; Heinrich, B.; Bourgogne, C.; Donnio, B.; Terazzi, E.; Bonnet, M.-E.; Stock, F.; Erbacher, P.; Bolcato-Bellemin, A.-L.; Douce, L. J. Am. Chem. Soc. 2009, 131, 13338–13346. doi:10.1021/ja903028f |
| 23. | Kantam, M. L.; Rao, B. P. C.; Choudary, B. M.; Reddy, R. S. Synlett 2006, 14, 2195–2198. doi:10.1055/s-2006-949615 |
© 2009 Fouchet et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)