
Abstract
Based on 5-(4-hydroxyphenyl)-2-octylpyrimidine 8, 5-phenylpyrimidine derivatives 3–7, 9 with different spacer chain lengths (C2 up to C6) and different terminal polar groups (Br, Cl, N3, OH, CN) were synthesized by etherification and nucleophilic substitution. The mesomorphic behaviour of these compounds was investigated by differential scanning calorimetry (DSC), polarizing optical microscopy (POM) and X-ray diffraction (WAXS and SAXS) and revealed smectic A mesophases for bromides, chlorides and azides 3, 4 and 6. For these compounds a maximum phase width was observed for the C5 spacer regardless of the terminal group, whereas the hydroxy- and cyano-substituted derivatives 5 and 7, respectively, were non mesomorphic and showed only melting transitions.
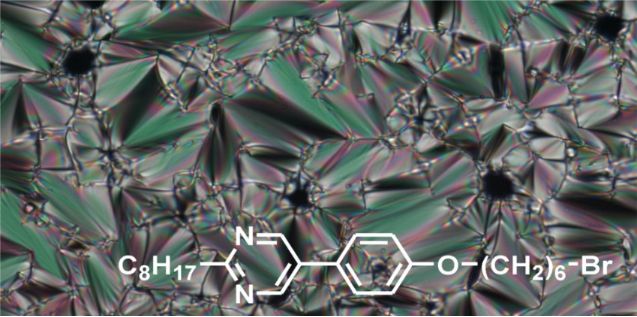
Graphical Abstract
Introduction
A tremendous amount of work has been done on calamitic liquid crystals, which has led to applications in the field of LC displays [1]. Among the large family of various calamitic mesogens 2-alkoxy-5-phenylpyrimidines 1 are prominent members due to the fact that the two nitrogen atoms increase the polarity of the rigid rod core structure (Scheme 1) as exemplified by the derivative 1a which displays a SmA phase between 45 °C and 71 °C, while the corresponding biphenyl derivative 2a with the same terminal alkyl chains does not have any liquid crystallinity [1].
![[1860-5397-5-63-i1]](/bjoc/content/inline/1860-5397-5-63-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Comparison of mesomorphic properties of 1a and 2a.
Scheme 1: Comparison of mesomorphic properties of 1a and 2a.
Whereas three regioisomeric phenylpyrimidines are possible, i.e. 4-, 5-, and 2-phenylpyrimidine, only the latter two are suitable for liquid crystals. Furthermore 5- and 2-phenylpyrimidines differ in their overall conformation. According to ab initio calculation by Barone [2], 2-phenylpyrimidine is almost planar, whereas 5-phenylpyrimidine has a twisted conformation with a dihedral angle of 43.1° [3]. The different conformations together with differences of the polarisation and dipole moment between 2- and 5-phenylpyrimidines also lead to different mesomorphic properties as was shown by Lemieux for phenylpyrimidines tethered to terminal trisiloxanes [4,5] and by Tschierske for dimeric phenylpyrimidines tethered to oligoethyleneglycol units [6].
We recently reported the synthesis of 1,1′-biisoquinolines tethered to calamitic subunits [7]. During these studies we discovered that the 5-phenylpyrimidine building block 3e already displayed a SmA mesophase. We thus wondered whether variation of the spacer chain lengths and terminal group X (Scheme 2) might have significant influence on the mesomorphism. The results of this study are discussed below.
![[1860-5397-5-63-i2]](/bjoc/content/inline/1860-5397-5-63-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Variation of spacer lengths and terminal group at 5-phenylpyrimidine.
Scheme 2: Variation of spacer lengths and terminal group at 5-phenylpyrimidine.
Results and Discussion
Syntheses: In order to obtain different series 3–7 the known 5-(4-hydroxyphenyl)-2-octylpyrimidine 8 [7-12] was used as starting material (Scheme 3).
![[1860-5397-5-63-i3]](/bjoc/content/inline/1860-5397-5-63-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of compounds 3–5, 9 and 11.
Scheme 3: Synthesis of compounds 3–5, 9 and 11.
Compound 8 was deprotonated with KOH in DMSO at room temperature for 10 min followed by addition of 1,ω-dibromoalkane. After 4 h, the reaction mixtures were purified and the desired bromides 3b–e were isolated in 23 up to 60% yield. When 1,3-dibromopropane was used, 27% of the elimination product 9 was isolated as byproduct. For comparison the corresponding 4-allyloxy-4′-octylbiphenyl 11 was prepared in 49% yield by allylation of 4-hydroxy-4′-octylbiphenyl. Compound 3a was obtained by etherification of 5-(4-hydroxyphenyl)-2-octylpyrimidine 8 using K2CO3 in MeCN under reflux for 12 h to yield 39%. The synthesis of series 4 with chloride as terminal group proceeded in a similar way by using 1,ω-dichloroalkanes giving 4a–e in 31–70% yield. Upon deprotonation of 8 under the conditions described above, followed by treatment with 5-bromopentanol or 6-bromohexanol, the hydroxy compounds 5d and 5e were isolated in 76% and 80% yield, respectively. To obtain the azides 6, bromides 3a–e were treated with NaN3 in DMF at 100 °C for 24 h and the products 6a–e were isolated in 74% up to quantitative yield (Scheme 4). In a similar manner, the cyanides 7 were prepared from the bromides 3. Treatment of the bromides 3c–e with KCN in EtOH/H2O at 110 °C for 12 h leads to the cyanides 7c–e in 72 to 97% yield.
![[1860-5397-5-63-i4]](/bjoc/content/inline/1860-5397-5-63-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of compounds 6 and 7.
Scheme 4: Synthesis of compounds 6 and 7.
Since the yields of 3b were limited due to side reactions like elimination, the respective cyanide 7b was prepared by deprotonation of hydroxy derivative 8 with NaH in DMF for 40 min at 0 °C followed by addition of 1-bromo-3-propionitrile at room temperature. After 12 h, the cyanide 7b was obtained in 33% yield.
Mesomorphic properties: Mesomorphic properties of compounds 3–10 were investigated by differential scanning calorimetry (DSC), polarizing optical microscopy (POM) and X-ray diffraction (WAXS and SAXS). The DSC results for bromides 3 are summarized in Table 1.
Table 1: Phase transition temperatures [°C] and enthalpies [kJ/mol] of compounds 3.a
| 3 | n | Cr1 | T | ΔH | Cr2 | T | ΔH | SmA | T | ΔH | I | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | 2 | • | 52 | 3.9 | • | 63 | 32.2 | - | - | - | • | 2. heating |
| • | 29 | −25.8 | - | - | - | • | 56 | −5.6 | • | 2. cooling | ||
| b | 3 | • | 46 | 13.0 | - | - | - | • | 52 | 1.9 | • | 2. heating |
| • | 35 | −13.9 | - | - | - | • | 52 | −3.2 | • | 2. cooling | ||
| c | 4 | • | 66 | 31.1 | - | - | - | - | - | - | • | 2. heating |
| • | 49 | −24.1 | - | - | - | • | 63 | −5.0 | • | 2. cooling | ||
| d | 5 | • | 40 | 17.6 | - | - | - | • | 57 | 5.1 | • | 2. heating |
| • | 24 | −16.7 | - | - | - | • | 60 | −5.3 | • | 2. cooling | ||
| e | 6 | • | 64 | 36.3 | - | - | - | - | - | - | • | 2. heating |
| • | 35 | −23.9 | - | - | - | • | 58 | −5.5 | • | 2. cooling | ||
aCr crystalline; SmA smectic A; I isotropic; • phase was observed; - phase was not observed. Heating and cooling rate: 10 K/min.
Whereas compounds 3a,c,e with even chain lengths of the spacer displayed monotropic SmA phases, compounds 3b,d with odd chain lengths displayed enantiotropic SmA phases. A typical DSC curve of derivative 3d with a pentyloxy spacer is shown in Figure 1.
![[1860-5397-5-63-1]](/bjoc/content/figures/1860-5397-5-63-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: DSC curve of compound 3d (heating/cooling rate 10 K/min).
Figure 1: DSC curve of compound 3d (heating/cooling rate 10 K/min).
On the third heating a melting transition at 40 °C into the SmA phase and a clearing transition at 57 °C could be observed. Subsequent cooling revealed an isotropic to SmA transition at 60 °C and a crystallization peak at 24 °C. The SmA to crystalline transition tends to strong supercooling in the order of 10–30 K (Table 1, Figure 1).
POM observation of 3a–e revealed fan-shaped textures typical of SmA phases. An illustrative example is depicted in Figure 2. The assignment of the SmA mesophases was further confirmed by XRD experiments (see the Supporting Information).
![[1860-5397-5-63-2]](/bjoc/content/figures/1860-5397-5-63-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Fan-shaped texture of 3e under crossed polarizers upon cooling from the isotropic liquid (magnification 200×): at 55 °C (cooling rate 5 K/min): smectic A phase.
Figure 2: Fan-shaped texture of 3e under crossed polarizers upon cooling from the isotropic liquid (magnifica...
It should be noted that the allyloxy-substituted byproduct 9 showed a smectic mesophase between 50 °C and 67 °C as well. In contrast, the corresponding 4-allyloxy-4′-octylbiphenyl 11 showed only isotropic melting at 92 °C. The DSC results of chlorides 4 are summarized in Table 2.
Table 2: Phase transition temperatures [°C] and enthalpies [kJ/mol] of compounds 4.a
| 4 | n | Cr | T | ΔH | SmA | T | ΔH | I | |
|---|---|---|---|---|---|---|---|---|---|
| a | 2 | • | 50 | 21.6 | • | 55 | 1.4 | • | 2. heating |
| • | 25 | −18.7 | • | 53 | −1.5 | • | 2. cooling | ||
| b | 3 | • | 37 | 11.9 | • | 53 | 3.9 | • | 2. heating |
| • | 23 | −11.8 | • | 58 | −4.2 | • | 2. cooling | ||
| c | 4 | • | 63 | 21.9 | • | 67 | 2.7 | • | 2. heating |
| • | 46 | −23.5 | • | 70 | −4.9 | • | 2. cooling | ||
| d | 5 | • | 42 | 15.7 | • | 56 | 5.0 | • | 2. heating |
| • | 27 | −17.0 | • | 62 | −4.8 | • | 2. cooling | ||
| e | 6 | • | 55 | 20.8 | • | 59 | 4.7 | • | 2. heating |
| • | 42 | −25.3 | • | 64 | −4.5 | • | 2. cooling | ||
aCr crystalline; SmA smectic A; I isotropic; • phase was observed; - phase was not observed. Heating and cooling rate: 10 K/min for 4a–d, 5 K/min for 4e.
All members 4a–e showed enantiotropic SmA phases. For compounds 4a,c,e with even numbered spacer lengths smaller mesophase widths were observed as compared to compounds 4b,d with odd numbered spacer lengths. Furthermore an odd–even effect of both melting and clearing points was found. A typical DSC curve which is shown in Figure 3 for chloride 4e with hexyloxy spacer, revealed a melting transition at 59 °C to the smectic A phase and a clearing transition at 66 °C upon a second heating. Upon the second cooling run an isotropic to SmA transition at 64 °C and a crystallization peak at 42 °C were observed.
![[1860-5397-5-63-3]](/bjoc/content/figures/1860-5397-5-63-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: DSC curve of compound 4e (heating/cooling rate 5 K/min).
Figure 3: DSC curve of compound 4e (heating/cooling rate 5 K/min).
POM investigation displayed fan-shaped and focal conic textures, as exemplified in Figure 4. XRD experiments proved the smectic phase.
![[1860-5397-5-63-4]](/bjoc/content/figures/1860-5397-5-63-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fan-shaped texture of compound 4d at 45 °C upon cooling from the isotropic liquid (cooling rate 1 K/min) (magnification 200×).
Figure 4: Fan-shaped texture of compound 4d at 45 °C upon cooling from the isotropic liquid (cooling rate 1 K...
In contrast to the bromides 3 and chlorides 4, the hydroxy and azide derivatives 5a,b and 7b–e were non mesomorphic and showed only melting transitions at 76 °C and 75 °C for compounds 5a,b and at 77 °C, 86 °C, 67 °C and 68 °C for the azides 7b–e, respectively (upon heating or cooling) in the DSC curve. Presumably, the higher polarity of the terminal hydroxy group with respect to the azido group, together with hydrogen bonding, inhibits mesophase formation. Next the azides 6 were investigated by DSC (Table 3).
Table 3: Phase transition temperatures [°C] and enthalpies [kJ/mol] of compounds 6.a
| 6 | n | Cr1 | T | ΔH | Cr2 | T | ΔH | SmA | T | ΔH | I | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | 2 | • | 48 | 30.6 | - | - | - | - | - | - | • | 2. heating |
| • | 44 | −31.4 | - | - | - | - | - | - | • | 2. cooling | ||
| b | 3 | • | 45 | 11.9 | - | - | - | • | 49 | 1.1 | • | 2. heating |
| • | 34 | −13.7 | - | - | - | • | 47 | −2.9 | • | 2. cooling | ||
| c | 4 | • | 42 | 25.3 | - | - | - | • | 60 | 5.0 | • | 2. heating |
| • | 28 | −17.8 | - | - | - | • | 63 | −5.3 | • | 2. cooling | ||
| d | 5 | • | 7 | 0.9 | • | 28 | 17.2 | • | 55 | 3.4 | • | 2. heating |
| • | 1 | −0.6 | • | 11 | −16.0 | • | 57 | −4.7 | • | 2. cooling | ||
| e | 6 | • | 41 | 26.5 | - | - | - | • | 53 | 6.1 | • | 2. heating |
| • | 20 | −22.2 | • | 25 | −0.5 | • | 58 | −5.9 | • | 2. cooling | ||
aCr crystalline; SmA smectic A; I isotropic; • phase was observed; - phase was not observed. Heating and cooling rate: 10 K/min.
Whereas compound 6a with an ethoxy spacer was non mesomorphic, enantiotropic SmA phases were detected for all other chain lengths 6b–e. Compound 6d showed an additional crystal to crystal transition. A typical DSC curve of derivative 6c is shown in Figure 5. POM revealed fan-shaped and focal conic texture, see for example Figure 6. Figure 7 and Figure 8 reveal that due to substantial supercooling for all spacer chain lengths and terminal groups the mesophases are smaller during the heating cycle as compared to the cooling cycle. The broadest mesophase was observed for the azide derivative 6d with ΔT = 27 °C upon heating and ΔT = 45 °C upon cooling. In comparison to the compounds with an azide as terminal group the halides (n = 2, 5, 6) have a lower tendency to supercooling. Whereas for azides 6 the broadest mesophase was observed for C5 spacer (6d), for chlorides 4 derivatives 4b and 4d with C3 and C5 spacer displayed similar mesophase width. For bromides 3 again the derivative 3d with C5 spacer showed the broadest mesophase.
![[1860-5397-5-63-5]](/bjoc/content/figures/1860-5397-5-63-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: DSC curve of compound 6c (heating/cooling rate 10 K/min).
Figure 5: DSC curve of compound 6c (heating/cooling rate 10 K/min).
![[1860-5397-5-63-6]](/bjoc/content/figures/1860-5397-5-63-6.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fan-shaped texture of compound 6e at 45 °C upon cooling from the isotropic liquid (cooling rate 10 K/min) (magnification 200×).
Figure 6: Fan-shaped texture of compound 6e at 45 °C upon cooling from the isotropic liquid (cooling rate 10 ...
![[1860-5397-5-63-7]](/bjoc/content/figures/1860-5397-5-63-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Comparison of the mesophase range ΔT for the different spacer lengths of compounds 3, 4 and 6: mesophase range upon heating (heating rate 10 K/min).
Figure 7: Comparison of the mesophase range ΔT for the different spacer lengths of compounds 3, 4 and 6: meso...
![[1860-5397-5-63-8]](/bjoc/content/figures/1860-5397-5-63-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Comparison of the mesophase range ΔT for the different spacer lengths of compounds 3, 4 and 6: mesophase range upon cooling (cooling rate 10 K/min).
Figure 8: Comparison of the mesophase range ΔT for the different spacer lengths of compounds 3, 4 and 6: meso...
From the X-ray data, the following model (Figure 9) of the layer structure is proposed. The d values obtained from the X-ray experiments fit with the molecular lengths derived from simple molecular modelling (Chem3D) [13]. For example, the XRD pattern of the azide derivative 6c results in a layer distance of 25.5 Å, whereas the calculated length of the molecule for the most elongated conformation is 26 Å, which is clear evidence for the presence of monolayers. This leads to the assumption that the molecules might be aligned antiparallel within each smectic layer (Figure 9). Packing the molecules in this array prevents close contacts between the polar regions of the rigid core and the terminal groups. The observed maximum phase width for the C5 spacer regardless of the terminal group suggests that for this chain length space filling is optimal and the terminal group X can be accommodated well between the alkyl chains. This model might also explain why the mesophase is lost with strongly polar or hydrogen bonding terminal groups such as cyanides and hydroxy derivatives.
![[1860-5397-5-63-9]](/bjoc/content/figures/1860-5397-5-63-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Proposed model for the layer structure.
Figure 9: Proposed model for the layer structure.
Conclusion
It has been shown that 5-phenylpyrimidine derivatives with terminal chloro-, bromo-, azido-, hydroxy- and cyano groups separated by an alkoxy spacer chain from the aromatic core were easily synthesised by nucleophilic substitution. Depending on the terminal group and the tether lengths the formation of smectic A mesophases was observed for the chloro-, bromo- and azido derivatives. Surprisingly, the strong latent dipole moment of hydroxy and cyano derivatives and the ability of hydroxy derivatives to form hydrogen bonds seems to completely suppress the formation of mesophases. Furthermore, the polar pyrimidine ring seems to play an important role in promoting liquid crystalline properties.
Experimental
General
Melting points were measured on a Mettler Toledo DSC822 and are uncorrected. NMR spectra were recorded on a Bruker Avance 300 and Avance 500 spectrometer. FT-IR spectra were recorded on a Bruker Vektor22 spectrometer with MKII Golden Gate Single Reflection Diamant ATR system. Mass spectra were recorded on a Finnigan MAT 95 and a Varian MAT 711 apparatus. X-Ray powder experiments were performed on a Bruker Nanostar; software: SAXS 4.1.26. The samples were kept in Hilgenberg glass capillaries of 0.7 mm outside diameter in a temperature-controlled heating stage (±1 °C). A monochromatic Cu-Kα1 beam (λ = 1.5405 Å) was obtained using a ceramic tube generator (1500 W) with cross-coupled Göbel-mirrors as the monochromator. The diffraction patterns were recorded on a real-time 2D-detector (HI-STAR, Bruker). The calibration of the patterns occurred with the powder pattern of Ag-Behenate. Differential scanning calorimetry (DSC) was performed using a Mettler Toledo DSC822, and polarizing optical microscopy (POM) using an Olympus BX50 polarizing microscope combined with a Linkam LTS350 hot stage and a Linkam TP93 central processor. Flash chromatography was performed using Kieselgel 60, 40–63 μm (Fluka). All solvents were dried, and reactions were performed in dried glassware. The used petroleum ether (PE) had a boiling range of 30–75 °C.
General procedure 1
To a solution of 5-(4-hydroxyphenyl)-2-octylpyrimidine 8 (852 mg, 3.00 mmol) in 4 mL DMSO was added powdered KOH (504 mg, 9.00 mmol). After stirring for 10 min at room temperature, the α,ω-dihaloalkane (or α-bromo-ω-alkanol respectively) (3.00 mmol) was added. Stirring was continued for 4 h followed by quenching with 20 mL H2O and 100 mL CH2Cl2. The organic layer was dried (Na2SO4) and the solvents were evaporated. Finally the crude product was purified by flash chromatography.
General procedure 2
A solution of bromide 3 (0.50 mmol) and NaN3 (81.0 mg, 1.25 mmol) in 15 mL DMF was stirred at 100 °C for 24 h. After cooling to room temperature, the reaction mixture was treated with 20 mL H2O and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried (Na2SO4), the solvent was evaporated and the crude product purified by flash chromatography.
General procedure 3
A solution of bromide 3 (0.50 mmol) and KCN (35.8 mg, 0.55 mmol) in 4 mL EtOH/H2O (3:1, v/v) was stirred at 110 °C for 12 h. After cooling to room temperature, 10 mL CH2Cl2 were added and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (1 × 10 mL) and dried (Na2SO4). Finally the solvent was evaporated and the crude product purified by flash chromatography.
5-[4-(6-Bromohexyloxy)phenyl]-2-octylpyrimidine (3e)
Prepared according to general procedure (1). Experiment: 284 mg (1.00 mmol) 5-(4-hydroxyphenyl)-2-octylpyrimidine 8, 160 μL (245 mg, 1.00 mmol) 1,6-dibromohexane, 168 mg (3.00 mmol) KOH. Flash chromatography (PE/EtOAc, 4:1, v/v; Rf = 0.86: PE/EtOAc, 1:1, v/v) gave 268 mg (0.60 mmol, 60%) of 3e as a colourless crystalline solid. DSC: Cr 35 °C [−23.9 kJ/mol] (SmA 58 °C [−5.5 kJ/mol]) I. 1H NMR (300 MHz, CDCl3): δ = 0.88 (t, 3H J = 6.9 Hz, CH3), 1.21–1.45 (m, 10H, CH2), 1.49–1.58 (m, 4H, CH2), 1.79–1.98 (m, 6H, CH2), 2.96–3.01 (m, 2H, 2-CH2), 3.44 (t, 2H, J = 6.6 Hz, CH2Br), 4.02 (t, 2H, J = 6.4 Hz, OCH2), 6.99–7.03 (m, 2H, 3′-H, 5′-H), 7.46–7.50 (m, 2H, 2′-H, 6′-H), 8.83 (s, 2H, 4-H, 6-H) ppm. 13C NMR (125 MHz, CDCl3): δ = 14.1 (CH3), 22.7, 25.3, 27.9, 28.9, 29.0, 29.2, 29.4, 31.9, 32.7, 33.8 (CH2), 39.2 (2-CH2), 67.9 (OCH2), 115.2 (C-3′, C-5′), 128.0 (C-2′, C-6′), 126.7, 130.8 (C-1′, C-5), 154.8 (C-4, C-6), 159.9 (C-4′), 169.5 (C-2) ppm. FT-IR (ATR): ![[Graphic 1]](/bjoc/content/inline/1860-5397-5-63-i5.svg?max-width=637&scale=1.18182) = 2916 (m), 2848 (m), 1586 (m), 1536 (m), 1515 (m), 1445 (s), 1247 (s), 1180 (m), 1116 (m), 1011 (m), 994 (m), 838 (s), 651 (m), 608 (m) cm−1. MS (EI, 70eV): m/z (%) = 446.1 (100) [M]+, 361.0 (20) [M−C6H13]+, 348.0 (78) [M + H−C7H15], 199.0 (10) 186.0 (28). C24H35BrN2O (447.45): calcd. C 64.42, H 7.88, N 6.26, Br 17.86; found C 64.46, H 7.88, N 6.14, Br 17.61.
= 2916 (m), 2848 (m), 1586 (m), 1536 (m), 1515 (m), 1445 (s), 1247 (s), 1180 (m), 1116 (m), 1011 (m), 994 (m), 838 (s), 651 (m), 608 (m) cm−1. MS (EI, 70eV): m/z (%) = 446.1 (100) [M]+, 361.0 (20) [M−C6H13]+, 348.0 (78) [M + H−C7H15], 199.0 (10) 186.0 (28). C24H35BrN2O (447.45): calcd. C 64.42, H 7.88, N 6.26, Br 17.86; found C 64.46, H 7.88, N 6.14, Br 17.61.
5-[4-(6-Chlorohexyloxy)phenyl]-2-octylpyrimidine (4e)
Prepared according to general procedure (1). Experiment: 85.0 mg (0.30 mmol) 5-(4-hydroxyphenyl)-2-octylpyrimidine 8, 45.0 μL (47.0 mg, 0.30 mmol) 1,6-dichlorohexane, 50.0 mg (0.90 mmol) KOH. Flash chromatography (PE/EtOAc, 5:1, v/v; Rf = 0.40: PE/EtOAc, 3:1, v/v) gave 74.0 mg (0.18 mmol, 60%) of 4e as a colourless crystalline solid. DSC: Cr 42 °C [−25.3 kJ/mol] SmA 64 °C [−4.5 kJ/mol] I. 1H NMR (300 MHz, CDCl3): δ = 0.85–0.90 (m, 3H, CH3), 1.23–1.44 (m, 10H, CH2), 1.50–1.55 (m, 4H, CH2), 1.78–1.90 (m, 6H, CH2), 2.96–3.01 (m, 2H, 2-CH2), 3.56 (t, 2H, J = 6.7 Hz, CH2Cl), 4.01 (t, 2H, J = 6.4 Hz, OCH2), 6.99–7.04 (m, 2H, 3′-H, 5′-H), 7.46–7.51 (m, 2H, 2′-H, 6′-H), 8.83 (s, 2H, 4-H, 6-H) ppm. 13C NMR (75 MHz, CDCl3): δ = 14.1 (CH3), 22.7, 25.4, 26.6, 28.8, 29.1, 29.2, 29.4, 31.9, 32.5 (CH2), 39.2 (2-CH2), 45.0 (CH2Cl), 67.9 (OCH2), 115.3 (C-3′, C-5′), 127.9 (C-2′, C-6′), 126.7, 130.8 (C-1′, C-5), 154.5 (C-4, C-6), 159.6 (C-4′), 169.6 (C-2) ppm. FT-IR (ATR): = 2917 (m), 2849 (m), 1587 (m), 1516 (m), 1446 (s), 1392 (m), 1287 (m), 1246 (s), 1181 (m), 1116 (m), 1029 (m), 994 (m), 941 (m), 838 (m), 721 (m) cm−1. MS (ESI): m/z = 403.3 [M + H]+, 367.3 [M–Cl]+. C24H35ClN2O (403.00): calcd. C 71.53, H 8.75, N 6.95, Cl 8.80; found C 71.38, H 8.63, N 6.81, Cl 8.93.
5-[4-(6-Hydroxyhexyloxy)phenyl]-2-octylpyrimidine (5e)
Prepared according to general procedure (1). Experiment: 568 mg (2.00 mmol) 5-(4-hydroxyphenyl)-2-octylpyrimidine 8, 270 μL (362 mg, 2.00 mmol) 6-bromohexane-1-ol, 336 mg (6.00 mmol) KOH. Flash chromatography (PE/EtOAc, 1:1, v/v; Rf = 0.29) gave 615 mg (1.60 mmol, 80%) of 5e as a colourless crystalline solid. Mp: 75 °C. 1H NMR (300 MHz, CDCl3): δ = 0.85–0.90 (m, 3H, CH3), 1.24–1.68 (m, 16H, CH2), 1.79–1.90 (m, 4H, CH2), 2.96–3.01 (m, 2H, 2-CH2), 3.68 (t, 2H, J = 6.5 Hz, CH2OH), 4.01 (t, 2H, J = 6.5 Hz, OCH2), 6.99–7.04 (m, 2H, 3′-H, 5′-H), 7.46–7.51 (m, 2H, 2′-H, 6′-H), 8.83 (s, 2H, 4-H, 6-H) ppm. 13C NMR (75 MHz, CDCl3): δ = 14.1 (CH3), 22.7, 25.6, 25.9, 28.9, 29.2, 29.5, 31.9, 32.7 (CH2), 39.2 (2-CH2), 62.8 (CH2OH), 68.0 (OCH2), 115.3 (C-3′, C-5′), 127.9 (C-2′, C-6′), 126.6, 130.8 (C-1’, C-5), 154.5 (C-4, C-6), 159.7 (C-4′), 169.6 (C-2) ppm. FT-IR (ATR): = 3303 (m, br), 2917 (s), 2848 (m), 1606 (m), 1586 (m), 1536 (m), 1516 (m), 1466 (m), 1445 (s), 1377 (m), 1291 (m), 1288 (m), 1248 (s), 1181 (m), 1118 (m), 1060 (m), 1007 (m), 994 (m), 918 (m), 838 (s), 707 (m), 652 (m) cm−1. MS (EI, 70eV): m/z (%) = 384.3 (100) [M]+, 299.2 (36), 286.2 (94). HRMS (ESI): m/z [M+H]+ calcd. for C24H37N2O2: 385.2850; found: 385.2856. C24H36N2O2 (384.55): calcd. C 74.96, H 9.44, N 7.28; found C 75.05, H 9.27, N 7.26.
5-[4-(6-Azidohexyloxy)phenyl]-2-octylpyrimidine (6e)
Prepared according to general procedure (2). Experiment: 224 mg (0.50 mmol)) bromide 3e, 81.0 mg (1.25 mmol) NaN3. Flash chromatography (PE/EtOAc, 2:1, v/v; Rf = 0.56) gave 205 mg (0.50 mmol, quant.) of 6e as a colourless crystalline solid. DSC: Cr1 20 °C [−22.2 kJ/mol] Cr2 25 °C [−0.5 kJ/mol] SmA 58 °C [−5.9 kJ/mol] I. 1H NMR (300 MHz, CDCl3): δ = 0.85–0.90 (m, 3H, CH3), 1.24–1.58 (m, 14H, CH2), 1.60–1.71 (m, 2 H, CH2), 1.78–1.91 (m, 4H, CH2), 2.97–3.01 (m, 2H, 2-CH2), 3.30 (t, 2H, J = 6.8 Hz, CH2N3), 4.02 (t, 2H, J = 6.4 Hz, OCH2), 7.00–7.04 (m, 2H, 3′-H, 5′-H), 7.47–7.51 (m, 2H, 2′-H, 6′-H), 8.83 (s, 2H, 4-H, 6-H) ppm. 13C NMR (75 MHz, CDCl3): δ = 14.1 (CH3), 22.7, 25.7, 26.5, 28.8, 28.9, 29.1, 29.2, 29.5, 31.9 (CH2), 39.2 (2-CH2), 51.4 (CH2N3), 67.9 (OCH2), 115.3 (C-3′, C-5′), 127.9 (C-2′, C-6′), 126.7, 130.8 (C-1’, C-5), 154.5 (C-4, C-6), 159.6 (C-4′), 169.6 (C-2) ppm. FT-IR (ATR): = 2921 (s), 2847 (m), 2091 (s), 1605 (m), 1587 (m), 1467 (m), 1446 (s), 1287 (m), 1246 (s), 1182 (m), 1032 (m), 993 (m), 836 (s), 654 (m) cm−1. MS (ESI): m/z = 432.3 [M + Na]+, 410.3 [M + H]+. C24H35N5O (409.58): calcd. C 70.38, H 8.61, N 17.10; found C 70.51, H 8.55, N 17.05.
5-[4-(6-Cyanohexyloxy)phenyl]-2-octylpyrimidine (7e)
Prepared according to general procedure (3). Experiment: 224 mg (0.50 mmol) bromide 3e, 36.0 mg (0.55 mmol) KCN. Flash chromatography (PE/EtOAc, 4:1, v/v; Rf = 0.23) gave 142 mg (0.36 mmol, 72%) of 7e as a colourless crystalline solid. Mp: 68 °C. 1H NMR (500 MHz, CDCl3): δ = 0.88 (t, 3H J = 6.9 Hz, CH3), 1.20–1.45 (m, 10H, CH2), 1.51–1.60 (m, 4H, CH2), 1.68–1.90 (m, 6H, CH2), 2.37 (t, 2H, J = 7.3 Hz, CH2CN), 2.96–3.01 (m, 2H, 2-CH2), 4.02 (t, 2H, J = 6.3 Hz, OCH2), 6.99–7.03 (m, 2H, 3′-H, 5′-H), 7.46–7.51 (m, 2H, 2′-H, 6′-H), 8.81 (s, 2H, 4-H, 6-H) ppm. 13C NMR (125 MHz, CDCl3): δ = 14.1 (CH3), 17.1, 22.7, 25.3, 25.4, 28.4, 28.9, 29.2, 29.5, 31.9 (CH2), 39.2 (2-CH2), 67.9 (OCH2), 115.4 (C-3′, C-5′), 119.7 (CN), 128.0 (C-2′, C-6′), 126.9, 130.8 (C-1’, C-5), 154.5 (C-4, C-6), 159.6 (C-4′), 169.7 (C-2) ppm. FT-IR (ATR): = 3035 (w), 2950 (m), 2920 (m), 2852 (m), 1608 (m) 1588 (m), 1541 (m), 1518 (m), 1440 (s), 1397 (m), 1245 (s), 1188 (s), 1048 (m), 996 (m), 836 (s), 738 (m), 706 (m), 652 (m), 556 (m) cm−1. MS (EI, 70eV): m/z (%) = 393.2 (72) [M]+, 350.2 (8), 308.2 (28), 395.2 (100), 185.1 (11). HRMS (ESI): m/z [M+H]+ calcd. for C25H36N3O: 394.2853; found: 394.2854. C25H35N3O (393.56): calcd. C 76.29, H 8.96, N 10.68; found C 76.15, H 8.93, N 10.51.
Supporting Information
Supporting information includes experimental and spectroscopic data for compounds 4a–d, 5d, 6a–d, 7a–d, 9, 11 and X-ray diffraction data.
| Supporting Information File 1: Analytical data of compounds 4a–d, 5d, 6a–d, 7a–d, 9, 11. | ||
| Format: PDF | Size: 1.1 MB | Download |
Acknowledgments
Generous financial support by the Deutsche Forschungsgemeinschaft, the Ministerium für Wissenschaft, Forschung und Kunst des Landes Baden-Württemberg (Landesgraduierten fellowship for Elisabeth Kapatsina), the Bundesministerium für Bildung und Forschung and the Fonds der Chemischen Industrie is gratefully acknowledged.
References
-
Goodby, J. W. In Handbook of Liquid Crystals; Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: Weinheim, Germany, 1998; Vol. 2A, pp 411–440.
Return to citation in text: [1] [2] -
Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664–675. doi:10.1063/1.475428
Return to citation in text: [1] -
Barone, V.; Commisso, L.; Lelj, F.; Russo, N. Tetrahedron 1985, 41, 1915–1918. doi:10.1016/S0040-4020(01)96554-8
Return to citation in text: [1] -
Li, L.; Jones, C. D.; Magolan, J.; Lemieux, R. P. J. Mater. Chem. 2007, 17, 2313–2318. doi:10.1039/b700972k
Return to citation in text: [1] -
Roberts, J. C.; Kapernaum, N.; Giesselmann, F.; Lemieux, R. P. J. Am. Chem. Soc. 2008, 130, 13842–13843. doi:10.1021/ja805672q
Return to citation in text: [1] -
Neumann, B.; Hegmann, T.; Wagner, C.; Ashton, P. R.; Wolf, R.; Tschierske, C. J. Mater. Chem. 2003, 13, 778–784. doi:10.1039/b210271d
Return to citation in text: [1] -
Kapatsina, E.; Lordon, M.; Baro, A.; Laschat, S. Synthesis 2008, 2551–2560. doi:10.1055/s-2008-1067184
Return to citation in text: [1] [2] -
Lloyd, D.; Reichardt, C.; Struthers, M. Liebigs Ann. Chem. 1986, 1368–1379. doi:10.1002/jlac.198619860807
Return to citation in text: [1] -
Dox, A. W. Org. Synth. 1941, 1, 5–6.
Return to citation in text: [1] -
Hooley, R. J.; Rebek, J. Org. Lett. 2007, 9, 1179–1182. doi:10.1021/ol062782s
Return to citation in text: [1] -
Sugita, S.-I.; Toda, S.; Yoshiyasu, T.; Teraji, T.; Murayama, A. Mol. Cryst. Liq. Cryst. 1994, 239, 113–122. doi:10.1080/10587259408047176
Return to citation in text: [1] -
Sugita, S.-I.; Takeno, H.; Teraji, T. Mol. Cryst. Liq. Cryst. 1991, 206, 139–146. doi:10.1080/00268949108037726
Return to citation in text: [1] -
Chem3D Pro 2008 software was used for molecular modelling.
Return to citation in text: [1]
| 1. | Goodby, J. W. In Handbook of Liquid Crystals; Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: Weinheim, Germany, 1998; Vol. 2A, pp 411–440. |
| 4. | Li, L.; Jones, C. D.; Magolan, J.; Lemieux, R. P. J. Mater. Chem. 2007, 17, 2313–2318. doi:10.1039/b700972k |
| 5. | Roberts, J. C.; Kapernaum, N.; Giesselmann, F.; Lemieux, R. P. J. Am. Chem. Soc. 2008, 130, 13842–13843. doi:10.1021/ja805672q |
| 3. | Barone, V.; Commisso, L.; Lelj, F.; Russo, N. Tetrahedron 1985, 41, 1915–1918. doi:10.1016/S0040-4020(01)96554-8 |
| 1. | Goodby, J. W. In Handbook of Liquid Crystals; Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: Weinheim, Germany, 1998; Vol. 2A, pp 411–440. |
| 7. | Kapatsina, E.; Lordon, M.; Baro, A.; Laschat, S. Synthesis 2008, 2551–2560. doi:10.1055/s-2008-1067184 |
| 8. | Lloyd, D.; Reichardt, C.; Struthers, M. Liebigs Ann. Chem. 1986, 1368–1379. doi:10.1002/jlac.198619860807 |
| 9. | Dox, A. W. Org. Synth. 1941, 1, 5–6. |
| 10. | Hooley, R. J.; Rebek, J. Org. Lett. 2007, 9, 1179–1182. doi:10.1021/ol062782s |
| 11. | Sugita, S.-I.; Toda, S.; Yoshiyasu, T.; Teraji, T.; Murayama, A. Mol. Cryst. Liq. Cryst. 1994, 239, 113–122. doi:10.1080/10587259408047176 |
| 12. | Sugita, S.-I.; Takeno, H.; Teraji, T. Mol. Cryst. Liq. Cryst. 1991, 206, 139–146. doi:10.1080/00268949108037726 |
| 7. | Kapatsina, E.; Lordon, M.; Baro, A.; Laschat, S. Synthesis 2008, 2551–2560. doi:10.1055/s-2008-1067184 |
| 6. | Neumann, B.; Hegmann, T.; Wagner, C.; Ashton, P. R.; Wolf, R.; Tschierske, C. J. Mater. Chem. 2003, 13, 778–784. doi:10.1039/b210271d |
© 2009 Starkulla et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
