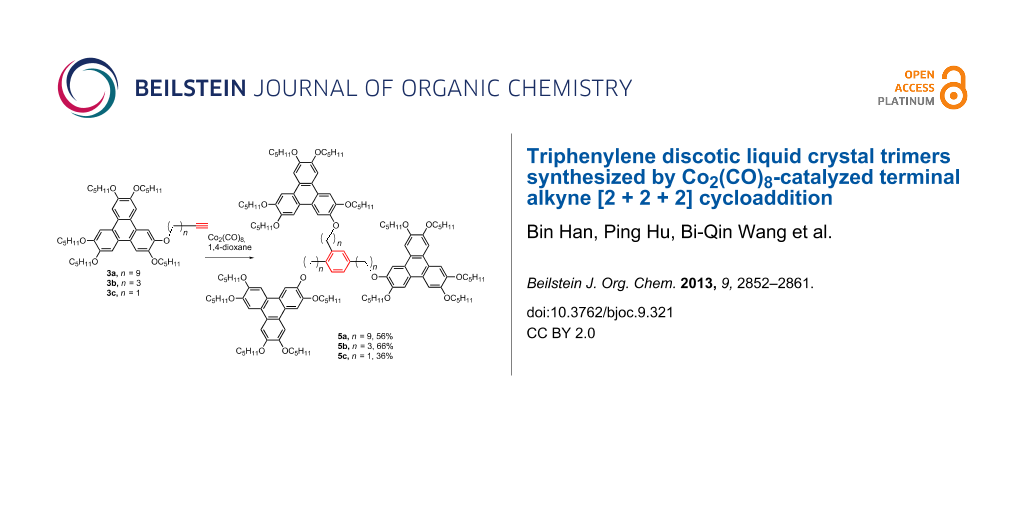
Abstract
The synthesis of star-shaped discotic liquid crystal trimers using Co2(CO)8-catalyzed terminal alkyne [2 + 2 + 2] cycloaddition reaction is reported. The trimers consist of three triphenylene discotic units linked to a central 1,2,4-trisubstituted benzene ring via flexible spacers. The trimers were synthesized in the yields up to 70% by mixing the monomers with 10 mol % of Co2(CO)8 as the catalyst in refluxing 1,4-dioxane. The liquid crystalline properties were investigated by using polarizing optical microscopy (POM), differential scanning calorimetry (DSC) and X-ray diffraction (XRD). Trimer 4 with an ester connecting group and a longer spacer exhibited a rectangular columnar mesophase, while 5b and 5c possessing an ether linkage and a shorter spacer display a hexagonal columnar mesophase. The connecting functional group and the length of the flexible spacer between the central benzene ring and the triphenylene units have pivotal influence on the mesomorphism.

Graphical Abstract
Introduction
Discotic liquid crystals (DLCs) with nematic phase have been commercially utilized in the liquid crystal display industry as optical compensating films for widening the view angles [1,2]. More interestingly, DLCs can self-organize into columnar mesophases with a high degree of order, and show fast unidirectional charge migration properties, and have been studied as soft organic semiconductors [3-12]. Solution-processed and ink-jet printing organic electronic devices based on liquid crystalline semiconductors are low-cost and thus are especially attractive to industry. Until now, DLCs have been explored as active materials applied in organic light-emitting diodes (OLEDs) [13-15], organic field-effect transistors (OFETs) [5-8,16,17], and organic photovoltaic solar cells (OPVs) [18-20].
The reported DLC oligomers [21-50] are limited compared with the low-molar-mass DLCs and polymeric DLC materials, due to the construction methods for the oligomers. However, DLC oligomers usually possess wider mesophase ranges than the monomers as the crystallization was prevented, due to the size of molecules is enlarged and molecular symmetry is lowered. However DLC oligomers exhibit higher charged carrier mobility than DLC polymers, the most important parameter in determining the device performance, as the discotic unites can still self-assemble to higher order through the π–π interaction. In addition, DLC oligomers similar to polymers can be processed on flexible substrates by spin-casting, screen printing, doctor-blading, ink-jet printing and roll-to-roll processing, so that these cost-effective deposition methods can be used to manufacture electronic devices. Therefore, the exploration of efficient synthetic methods for the DLC oligomers and studying the properties of them are fundamental.
The construction methods of DLC oligomers can be divided into conventional organic reactions and transition metal-catalyzed synthetic methods. The use of transition metal-mediated reactions for constructing new organic functional materials is more efficient and therefore attractive. Our group has embarked upon a program to use new organic synthetic methods to prepare novel types of DLC materials and to study the relationship between their molecular structures and the mesomorphic properties [46-50]. We have reported that Cu(I)-catalyzed alkyne–azide click reactions are emerging as an efficient method for the synthesis of discotic oligomers [46-50].
The transition metal-catalyzed [2 + 2 + 2] cycloaddition of three alkynes for the synthesis of polysubstituted benzene derivatives was studied due to its high efficiency and atomic efficiency [51]. The use of Co2(CO)8 as a catalyst has been extensively applied to the synthesis of hexabenzocoronene DLCs through diarylethyne cyclotrimerization [51].
In this paper, we report four star-shaped DLC trimers with triphenylene discotic units by using a Co2(CO)8-catalyzed terminal alkynes [2 + 2 + 2] cycloaddition, and the trimers exhibit ordered rectangular (Colro) and hexagonal columnar mesophases (Colho). Furthermore, the structure-mesomorphic property relationship is discussed. The synthetic route is shown in Scheme 1 and Scheme 2.
![[1860-5397-9-321-i1]](/bjoc/content/inline/1860-5397-9-321-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of star-shaped triphenylene discotic liquid crystalline trimer 4.
Scheme 1: Synthesis of star-shaped triphenylene discotic liquid crystalline trimer 4.
![[1860-5397-9-321-i2]](/bjoc/content/inline/1860-5397-9-321-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of star-shaped triphenylene discotic liquid crystalline trimers 5a–c.
Scheme 2: Synthesis of star-shaped triphenylene discotic liquid crystalline trimers 5a–c.
Results and Discussion
Synthesis and characterization
We synthesized the key intermediate 1, 2-hydroxy-3,6,7,10,11-pentakis(pentyloxy)triphenylene, according to a simplified one-pot method [52]. The undec-10-yn-1-yl p-toluenesulfonate was prepared by LiAlH4 reduction of the acid and tosylation of the alcohol [53,54]. Then monomers 2 and 3a–c were synthesized by the direct esterification or etherification reaction between phenol 1 and 10-undecynoic acid, undec-10-yn-1-yl p-toluenesulfonate, 5-chloro-1-pentyne, propargyl bromide, respectively.
The star-shaped DLC trimers with triphenylene discogens, 4 and 5a–c, were synthesized in yields of 36–71% by the self-trimerization of monomer 2 or 3a–c catalyzed by using 10 mol % of Co2(CO)8 in refluxing 1,4-dioxane. Trimer 4, 5a and 5b were prepared in moderate yields, and the obvious lower synthetic yield of 5c might be caused by its shorter spacer and bigger steric hindrance. Considering the size of the trimers, we were satisfied with the preliminary synthetic yields, and did not further optimize the reaction conditions.
Two isomers were obtained in the trimerization of mono-substituted alkynes, R-C≡CH: 1,2,4- and 1,3,5-trialkylbenzene. For the DLC trimers, the isomers could not be separated by thin-layer chromatography and column chromatography, and even high performance liquid chromatography (HPLC). However, the benzenes with three substituents, a 1,2,4- or 1,3,5-trisubstituted pattern, can be characterized by 1H NMR spectroscopy [55,56]. According to this method, we find that the 1H NMR peak of the 1,3,5-trisubstituted benzene isomer 4 appears at 6.83 ppm, for 5a at 6.81 ppm, and 7.03 ppm for 5b. There was no signal for the 1,3,5-trisubstituted isomer for 5c. The 1H NMR peak area integration results showed that for 4, 5a and 5b, the 1,2,4-trisubstituted benzenes were present in more than 95% and 1,3,5-trisubstituted benzene isomers were less than 5%. For 5c, the symmetric isomer of the 1,3,5-trisubstituted benzene was not detected, and the yield of the 1,2,4-trisubstituted isomer was almost quantitative. Therefore, we came to the conclusion that this synthetic method and the following purification procedures supplied the 1,2,4-trisubstituted benzene-cored DLC oligomers.
Mesomorphism
POM and DSC
Initially, we studied the mesomorphic properties of the monomers and trimers by using polarizing optical microscopy (POM) and differential scanning calorimetry (DSC). The POM results of the monomers and trimers are summarized in Figure 1 and Figure 2, respectively. The DSC traces are shown in Figure 3 and the phase transition data are summarized in Table 1.
![[1860-5397-9-321-1]](/bjoc/content/figures/1860-5397-9-321-1.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Optical photomicrographs of the triphenylene DLC monomers. (A) 2 at 40 °C; (B) 3a at 45 °C; (C) 3b at 62 °C; (D) 3c at 67 °C.
Figure 1: Optical photomicrographs of the triphenylene DLC monomers. (A) 2 at 40 °C; (B) 3a at 45 °C; (C) 3b ...
![[1860-5397-9-321-2]](/bjoc/content/figures/1860-5397-9-321-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Optical photomicrographs of the triphenylene DLC trimers. (A) 4 at 70 °C; (B) 5b at 85 °C; (C) 5c at 100 °C; (D) 5c at 75 °C.
Figure 2: Optical photomicrographs of the triphenylene DLC trimers. (A) 4 at 70 °C; (B) 5b at 85 °C; (C) 5c a...
![[1860-5397-9-321-3]](/bjoc/content/figures/1860-5397-9-321-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The DSC traces of the triphenylene DLC monomers and trimers. (A) 2nd heating traces; (B) 1st cooling traces. Scanning rate 10 K/min.
Figure 3: The DSC traces of the triphenylene DLC monomers and trimers. (A) 2nd heating traces; (B) 1st coolin...
Table 1: Thermotropic phase-transition behavior of the triphenylene DLC monomers and trimers. (Heating and cooling rate of 10 K/min.)a
| Compd. | 2nd heating | 1st cooling |
|---|---|---|
| Transition temperature (°C) and enthalpy change (ΔH, kJ/mol) | Transition temperature (°C) and enthalpy change (ΔH, kJ/mol) | |
| TP(OC5H11)6 | 69 Colho 122 | |
| 2b | Col 111 (9.8) Iso | Iso 110 (9.8) Col |
| 3a | Cr 41 (47.8) Col 60 (4.8) Iso | Iso 59 (5.1) Col 11 (43.0) Cr |
| 3bb | Cr 69 (44.2) Col 117 (10.3) Iso | Iso 117 (10.4) Col 19 (26.5) Cr |
| 3c | Cr 80 (67.5) Col 121 (11.4) Iso | Iso 121 (11.1) Col 23 (46.7) Cr |
| 4 | Col 111 (9.5) Iso | Iso 100 (4.7) Col |
| 5a | Cr 25 (14.3) Iso | Iso 15 (14.6) Cr |
| 5b | Col 106 (10.9) Iso | Iso 104 (10.5) Col |
| 5c | Col 125 (19.1) Iso | Iso 101 (10.4) Col |
aCr, crystal state; Col, columnar phase; Iso, isotropic liquid. bMonomer 2 [52] and 3b [57] were reported and the mesomorphism is comparable.
For the mesomorphism of the functionalized triphenylene monomers, compound 2 [52] and 3b [57] have been reported, 3a and 3c are new. They all display typical optical textures with homeotropic alignment behavior of the hexagonal columnar (Colh) mesophase (Figure 1). The monomers display different phase-transition temperatures related to the connecting functional group and the length of the chain. Compound 2 displayed a Col phase at room temperature with a clearing point of 111 °C (for the heating run), and did not crystallize even when cooled to −50 °C. Monomer 3a exhibited a narrow columnar mesophase range between 41 °C to 60 °C for heating, and between 59 °C to 11 °C for cooling. Comparing the phase-transition temperatures of 2 and 3a, it was found that the ester connecting group has the effect of lowering the melting point and rising the clearing point. Compared with the symmetric discogen of 2,3,6,7,10,11-hexakis(pentyloxy)triphenylene (C5OTP) which possesses a Col phase between 69–122 °C [58], monomer 2 displays a lower melting point and 3a exhibits both lowered melting and clearing points. The ester connecting group of 2 increases its dipole–dipole interactions in the intermolecular columnar stacking and stabilizes the Col mesophase. The results here are in agreement with the earlier works of Wendorff, Ringsdorf and Spiess [59-64].
Both 3b and 3c having slightly lowered molecular symmetry compared to the parent compound C5OTP, exhibited a Col mesophase between 69–117 °C and 80–121 °C, respectively.
Figure 2 shows the POM results for the trimers. Trimer 4 self-assembled into small-sized domains which were independent of the temperature and the cooling rate, and displayed a clearing point at 110 °C on heating, and on the cooling run the Col phase appeared at 100 °C. The crystallization did not occur for 4 even when it was cooled to −50 °C at the rate of 10 K/min.
Trimer 5a did not show mesomorphism as indicated both from the results of the POM and the DSC. The DSC curves of 5a showed only one phase transition peak at 15 °C on the first cooling run and at 25 °C on the second heating run. From the POM observations, we noted that the peak represents the crystal to isotropic liquid transition (Cr→Iso) and the reversed Iso→Cr transition.
Both trimer 5b and 5c exhibited a similar mesophase behavior to trimer 4: There is only one phase transition peak on the first cooling process and the second heating run. For 5b, the Col→Iso transition occurred at 106 °C on heating, and Iso→Col appeared at 104 °C on cooling. For 5c, the Col→Iso transition occurred at 125 °C on heating, and the reversed transition at 101 °C on cooling. No crystallization was observed for either 5b or 5c.
XRD results
Figure 4 depicts the X-ray diffraction patterns of 4, 5b and 5c at room temperature cooling from the isotropic liquid. Both 5b and 5c show a strong diffraction peak in the small-angle region (2θ = 3.8° or 4.5°), a broad halo peak of the alkyl chain at ca. 18°, and a core–core distance peak at 3.5–3.7 Å. Considering these XRD results together with their homeotropic alignment behavior displayed by the POM results shown in Figure 2C and Figure 2D, we assigned the mesophase of 5b and 5c as the Colho phase.
![[1860-5397-9-321-4]](/bjoc/content/figures/1860-5397-9-321-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Powder X-ray diffraction patterns of the DLC trimmers 4, 5b and 5c at room temperature.
Figure 4: Powder X-ray diffraction patterns of the DLC trimmers 4, 5b and 5c at room temperature.
However, 4 exhibited a different XRD pattern from that of 5b and 5c: two strong diffraction peaks in the small-angle region (2θ = 3.7° and 4.7°) with a d value of 23.78 Å and 18.80 Å, respectively. We assigned the mesophase of 4 as the rectangular columnar phase (Colro), which was further confirmed by the temperature-dependent XRD results (Figure 5), and the non-homeotropic alignment behavior of the POM texture (Figure 2A). The lattice parameters of the discotic Col mesophase of the trimers are summarized in Table 2. Trimer 4 displayed a smaller intracolumnar core–core distance of 3.57Å than that of 5b and 5c, due to the stronger π–π interactions between the triphenylene discogens with mono-ester group. The columnar parameter values of the trimers decreased with the spacer length shortened. So we deduced that the benzene cores were among the alkyl chains in the columnar stacking of the trimers.
![[1860-5397-9-321-5]](/bjoc/content/figures/1860-5397-9-321-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: X-ray diffraction profiles of 4 at different temperatures.
Figure 5: X-ray diffraction profiles of 4 at different temperatures.
Table 2: Columnar mesophase parameters of the DLC trimers.
| Compd. | hkl | dhkl (Å) | lattice parameters (Å) |
|---|---|---|---|
| 4 | 200 | 23.78 | a = 47.56 (Colro) |
| (25 °C) | 110 | 18.80 | b = 20.47 |
| alkyl halo | 4.70 | ||
| 001 | 3.57 | ||
| 5b | 100 | 22.97 | 26.52 (Colho) |
| (25 °C) | alkyl halo | 4.87 | |
| 001 | 3.75 | ||
| 5c | 100 | 19.70 | 22.75 (Colho) |
| (25 °C) | alkyl halo | 4.87 | |
| 001 | 3.65 | ||
Further considering the relationship between the molecular structures of the trimers and the mesomorphism, we noted first the mesophase disappearance of 5a, which was synthesized by trimerization of liquid crystalline 3a. However, trimer 4 has displayed a stable Colro mesophase while 5a did not. Both 4 and 5a possess the same length spacer, the ester-linker of 4 has a stronger polarity than the ether-linker of 5a. The isotropic point of 4 (111 °C) is higher than that of 5a (25 °C). More importantly, 4 is a room temperature liquid crystal and 5a has not shown a mesophase. Compared with 5a, both 5b and 5c have shorter spacers and have displayed a stable Colho mesophase over a wide temperature range.
Our study has demonstrated that for the star-shaped DLC trimers, both the length of spacer and the connecting linker group to the discotic units play a crucial role in the formation and stabilization of the discotic columnar mesophase.
Conclusion
The synthesis and mesomorphism of two new mono-functionalized triphenylene discotic monomers and four discotic trimers is reported. The trimers have been successfully synthesized for the first time by using a Co2(CO)8-catalyzed terminal alkyne [2 + 2 + 2] cycloaddition reaction in moderate yields. Three of the four 1,2,4-trisubstituted benzene-cored discotic trimers have shown stable Colho and Colro mesophases and wide mesophase ranges including room temperature. The connecting linker group to the triphenylene and the spacer length to the central benzene core have important impacts on the supramolecular packing and phase-transition temperature. We anticipate that this Co2(CO)8-catalyzed synthetic method can be used for the synthesis of liquid crystalline polymer networks.
Experimental
Instruments and conditions: See Supporting Information File 1.
Chemical reagents and starting materials
10-undecynoic acid, 5-chloro-1-pentyne, propargyl bromide and Co2(CO)8 were purchased from Alfa Aesar (Tianjin, China). All the other reagents and solvents were commercial products and used without further purification unless otherwise noted. 10-Undecyn-1-yl p-toluenesulfonate [53,54], 2-hydroxy-3,6,7,10,11-pentakis(pentyloxy)triphenylene [52] were synthesized as described in the reported methods.
Monomer 2, C18H6(OC5H11)5(OOCC8H16-C≡CH): Monomer 2 was prepared similar to the reported method [52]. The mixture of 10-undecynoic acid (218 mg, 1.2 mmol), dicyclohexylcarbodiimide (DCC, 247 mg, 1.2 mmol), 4-N,N-dimethylaminopyridine (DMAP, 67 mg), and 2-hydroxy-3,6,7,10,11-pentakis(pentyloxy)triphenylene (1, 674 mg, 1.0 mmol) in CH2Cl2 (20 mL) was stirred at room temperature for 6 h under a nitrogen atmosphere. After the reaction was finished, water was added to the mixture and then extracted with CH2Cl2. The extract was dried with MgSO4 and the solvent was distilled off using a rotary evaporator. The crude product was purified by column chromatography on silica gel eluted with CH2Cl2/hexane (3:2 in volume). The product was re-crystallized from ethanol affording a white solid (543 mg, 0.65 mmol, 65%). 1H NMR (CDCl3, TMS, 400 MHz) δ 8.06 (s, 1H, ArH), 7.85–7.77 (m, 5H, ArH), 4.26–4.19 (m, 10H, CH2), 2.67 (t, J = 7.6 Hz, 2H, CH2), 2.20 (td, J = 7.2 Hz, J = 2.8 Hz, 2H, CH2), 1.96–1.87 (m, 13H, CH2, C≡CH), 1.57–1.39 (m, 30H, CH2), 0.99–0.97 (t, J = 7.2 Hz, 15H, CH3).
Monomer 3a, C18H6(OC5H11)5(OC9H18-C≡CH): The mixture of 10-undecyn-1-yl p-toluenesulfonate (387 mg, 1.2 mmol), K2CO3 (345 mg, 2.5 mmol), and 2-hydroxy-3,6,7,10,11-pentakis(pentyloxy)triphenylene (1, 74 mg, 1.0 mmol) in DMF (15 mL) was stirred at 80 °C for 24 h under N2. The mixture was cooled to room temperature, and 3 M HCl was added drop-wise until the mixture was acidic. The organic phase was extracted with CH2Cl2 and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to afford the crude product, which was purified by column chromatography on silica gel eluted with CH2Cl2/hexane. The product was re-crystallized from ethanol to afford a light yellow solid (626 mg, 0.76 mmol, 76%). 1H NMR (CDCl3, TMS, 400 M) δ 7.84 (s, 6H, ArH), 4.23 (t, J = 6.4 Hz, 12H, CH2), 2.21–2.17 (m, 2H, CH2) 1.99–1.92 (m, 13H, CH2, C≡CH), 1.60–1.50 (m, 14H, CH2), 1.49–1.36 (m, 18H, CH2), 0.99–0.96 (m, 15H, CH3); 13C NMR (CDCl3, 100 MHz) δ 148.9, 123.6, 107.2, 84.7, 69.6, 68.1, 29.5, 29.1, 28.8, 28.5, 28.4, 26.2, 22.6, 18.4, 14.2; anal. calcd for C54H80O6: C, 78.60; H, 9.77; found: C, 78.36; H, 9.68.
Monomer 3b, C18H6(OC5H11)5(OC3H6-C≡CH): Monomer 3b was synthesized in a similar way to literature [57]. A mixture of 5-chloro-1-pentyne (230 mg, 2.25 mmol), K2CO3 (621 mg, 4.50 mmol), and 2-hydroxy-3,6,7,10,11-pentakis(pentyloxy)triphenylene (1, 1011 mg, 1.5 mmol) in DMF (20 mL) was stirred at 80 °C for 24 h under N2. The crude product was purified through column chromatography and a white solid was obtained (959 mg, 1.3 mmol, 86%). 1H NMR (CDCl3, TMS, 400 MHz) δ 7.87–7.84 (m, 6H, ArH), 4.34 (t, J = 6.0 Hz, 2H, CH2), 4.23 (t, J = 6.4 Hz, 10H, CH2), 2.54 (td, J = 7.2 Hz, J = 2.8 Hz, 2H, CH2), 2.18–2.14 (m, 2H, CH2), 2.00 (t, J = 2.8 Hz, 1H, C≡CH), 1.99–1.92 (m, 10H, CH2), 1.60–1.50 (m, 10H, CH2), 1.49–1.41 (m, 10H, CH2), 0.98 (t, J = 7.2 Hz, 15H, CH3); 13C NMR (CDCl3, 100 MHz), δ 149.0, 148.9, 148.6, 123.8, 123.6, 123.5, 107.6, 107.2, 107.1, 83.7, 69.6, 68.9, 68.0, 29.1, 28.4, 22.6, 15.3, 14.1; anal. calcd for C48H68O6: C, 77.80; H, 9.25; found: C, 77.70; H, 9.33.
Monomer 3c, C18H6(OC5H11)5(OCH2-C≡CH): A mixture of propargyl bromide (268 mg, 2.25 mmol), K2CO3 (621 mg, 4.5 mmol), and 2-hydroxy-3,6,7,10,11-pentakis(pentyloxy)triphenylene (1, 1011 mg, 1.5 mmol) in DMF (20 mL) was stirred at 80 °C for 24 h under N2. The crude product was purified with column chromatography and a white solid was collected (1013 mg, 1.42 mmol, 95%). 1H NMR (CDCl3, TMS, 400 MHz) δ 8.08 (s, 1 H, ArH), 7.85 (s, 1H, ArH), 7.83 (s, 4H, ArH), 4.97 (d, J = 1.2 Hz, 2H, CH2), 4.27–4.22 (m, 10H, CH2), 2.58 (t, J = 2.4 Hz, 1H, C≡CH), 1.99–1.92 (m, 10H, CH2), 1.60–1.53 (m, 10H, CH2), 1.50–1.43 (m, 10H, CH2), 0.98 (t, J = 7.6Hz, 15H, CH3); 13C NMR (CDCl3, 100 MHz) δ 149.2, 149.0, 148.8, 146.7, 124.7, 123.9, 123.5, 123.4, 123.3, 123.2, 109.8, 107.4, 107.2, 107.0, 106.7, 106.5, 78.9, 75.9, 69.7, 69.5, 69.3, 57.8, 29.1, 29.0, 28.4, 28.3, 22.6, 14.1; anal. calcd for C46H64O6: C, 77.49; H, 9.05; found: C, 77.41; H, 9.00.
Trimer 4: A mixture of 2 (252 mg, 0.3 mmol) and Co2(CO)8 (10 mg, 0.03 mmol) in dioxane (20 mL) was stirred under reflux for 24 h under an argon atmosphere, then cooled to room temperature. The organic phase was extracted with CH2Cl2, dried over MgSO4 and the solvent was removed. The crude product was purified by column chromatography on silica gel eluted with CH2Cl2/hexane (3:1 in volume), and then re-crystallized from ethanol affording a white solid (178 mg, 0.21 mmol, 71%). 1H NMR (CDCl3, TMS, 600 MHz) δ 8.02–8.00 (m, 3H, ArH), 7.81–7.71 (m, 15H, ArH), 7.06 (d, J = 3.6 Hz, 1H, ArH), 6.97 (s, 1H, ArH), 6.95 (d, J = 3.9 Hz, 1H, ArH), 6.83 (the 1,3,5-trisubstituted benzene core), 4.23–4.18 (m, 30H, CH2), 2.66 (t, J = 7.5 Hz, 6H, CH2), 2.61–2.55 (m, 6H, CH2), 1.97–1.81 (m, 36H, CH2), 1.66–1.34 (m, 90H, CH2), 0.99–0.95 (m, 45H, CH3); 13C NMR (CDCl3, 100 MHz) δ 172.1, 149.5, 149.3, 149.0, 148.7, 148.6, 140.3, 140.2, 139.6, 137.6, 129.2, 129.0, 127.8, 125.8, 124.5, 123.3, 123.0, 122.9, 116.6, 107.7, 107.0, 106.6, 106.3, 105.8, 69.7, 69.3, 69.1, 68.7, 35.7, 34.2, 32.9, 32.4, 31.7, 31.5, 30.0, 29.9, 29.6, 29.5, 29.4, 29.2, 29.1, 28.4, 28.3, 25.2, 22.6, 14.2; anal. calcd for C162H234O21: C, 77.29; H, 9.37; found: C, 77.61; H, 9.39.
Trimer 5a: Trimer 5a was synthesized by the same method as 4, which afforded a colorless oily product (186 mg, 0.23 mmol, 56%). 1H NMR (CDCl3, TMS, 600 MHz) δ 7.82 (s, 18H, ArH), 7.04 (d, J = 3.9 Hz, 1H, ArH), 6.95 (s, 1H, ArH), 6.93 (d, J = 3.9 Hz, 1H, ArH), 6.81 (the 1,3,5-trisubstituted benzene core), 4.23–4.21 (m, 36H, CH2), 2.58–2.52 (m, 6H, CH2), 1.97–1.91 (m, 36H, CH2), 1.58–1.35 (m, 96H, CH2), 0.98–0.95 (m, 45H, CH3); 13C NMR (CDCl3, 100 MHz) δ 149.1, 149.0, 148.9, 148.8, 148,7, 140.3, 140.1, 137.6, 129.2, 128.9, 125.7, 123.7, 123.6, 123.5, 123.4, 107.3, 107.2, 107.1, 107.0, 69.6, 35.7, 32.8, 32.4, 31.7, 31.5, 31.4, 30.0, 29.9, 29.7, 29.6, 29.5, 29.1, 28.4, 26.3, 22.6, 14.1; anal. calcd for C162H240O18: C, 78.60; H, 9.77; found: C, 78.55; H, 9.79.
Trimer 5b: 5b was synthesized by the same method as 4, which afforded a white solid (195 mg, 0.26 mmol, 66%). 1H NMR (CDCl3, TMS, 600 MHz) δ 7.82–7.73 (m, 18H, ArH), 7.22 (d, J = 3.9 Hz, 1H, ArH), 7.18 (s, 1H, ArH), 7.09 (dd, J = 3.9 Hz, J = 0.9 Hz, 1H, ArH), 7.03 (the 1,3,5-trisubstituted benzene core), 4.28–4.13 (m, 36H, CH2), 3.0 (t, J = 7.9 Hz, 4H, CH2), 2.88 (t, J = 8.0 Hz, 2H, CH2), 2.28–2.18 (m, 6H, CH2), 1.97–1.85 (m, 30H, CH2), 1.59–1.35 (m, 60H, CH2), 0.99–0.87 (m, 45H, CH3); 13C NMR (CDCl3, 100 MHz) δ 148.9, 148.8, 148.7, 148.6, 139.8, 139.7, 137.2, 129.8, 129.6, 126.4, 123.6, 123.5, 123.4, 107.2, 107.1, 106.9, 106.8, 69.6, 69.5, 69.4, 69.3, 68.7, 68.5, 32.0, 31.9, 31.3, 31.2, 31.1, 29.7, 29.2, 29.0, 28.6, 28.4, 22.6, 14.1; anal. calcd for C144H204O18: C, 77.80; H, 9.25; found: C, 77.83; H, 9.26.
Trimer 5c: Trimer 5c was synthesized by the same method as 4, which afforded a white solid (117 mg, 0.16 mmol, 36%). 1H NMR (CDCl3, TMS, 600 MHz) δ 7.95–7.60 (m, 21H, ArH), 5.65 (s, 4H, CH2), 5.37 (s, 2H, CH2), 4.24–4.12 (m, 22H, CH2), 4.07–4.03 (m, 4H, CH2), 3.97–3.94 (m, 4H, CH2), 1.98–1.86 (m, 22H, CH2), 1.80–1.73 (m, 4H, CH2), 1.70–1.65 (m, 4H, CH2), 1.59–1.37 (m, 44H, CH2), 1.36–1.30 (m, 8H, CH2), 1.28–1.16 (m, 8H, CH2), 1.01–0.94 (m, 27H, CH3), 0.93–0.90 (m, 6H, CH3), 0.83–0.78 (m, 9H, CH3), 0.75 (t, J = 7.3 Hz, 3H, CH3); 13C NMR (CDCl3, 100 MHz) δ 149.0, 148.9, 148.8, 137.6, 136.1, 135.4, 129.2, 128.1, 127.2, 124.0, 123.3, 108.7, 107.2, 107.0, 106.9, 106.6, 106.5, 105.9, 71.6, 70.5, 70.2, 69.7, 69.6, 69.5, 69.4, 69.1, 69.0, 29.2, 29.1, 29.0, 28.4, 28.3, 22.6, 14.1; anal. calcd for C138H192O18: C, 77.49; H, 9.05; found: C, 77.77; H, 9.09.
Supporting Information
| Supporting Information File 1: Characterization instruments and methods. 1H NMR spectra and 13C NMR spectra for the monomers and trimers. | ||
| Format: PDF | Size: 615.2 KB | Download |
References
-
Bushby, R. J.; Kawata, K. Liq. Cryst. 2011, 38, 1415–1426. doi:10.1080/02678292.2011.603262
Return to citation in text: [1] -
Kawata, K. Chem. Rec. 2002, 2, 59–80. doi:10.1002/tcr.10015
Return to citation in text: [1] -
Adam, D.; Schuhmacher, P.; Simmerer, J.; Häussling, L.; Siemensmeyer, K.; Etzbachi, K. H.; Ringsdorf, H.; Haarer, D. Nature 1994, 371, 141–143. doi:10.1038/371141a0
Return to citation in text: [1] -
Laschat, S.; Baro, A.; Steinke, N.; Giesselmann, F.; Hägele, C.; Scalia, G.; Judele, R.; Kapatsina, E.; Sauer, S.; Schreivogel, A.; Tosoni, M. Angew. Chem., Int. Ed. 2007, 46, 4832–4887. doi:10.1002/anie.200604203
Return to citation in text: [1] -
Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c
Return to citation in text: [1] [2] -
Shimizu, Y.; Oikawa, K.; Nakayama, K.; Guillon, D. J. Mater. Chem. 2007, 17, 4223–4229. doi:10.1039/b705534j
Return to citation in text: [1] [2] -
Funahashi, M. Polym. J. 2009, 41, 459–469. doi:10.1295/polymj.PJ2008324
Return to citation in text: [1] [2] -
O’Neill, M.; Kelly, S. M. Adv. Mater. 2011, 23, 566–584. doi:10.1002/adma.201002884
Return to citation in text: [1] [2] -
Kaafarani, B. R. Chem. Mater. 2011, 23, 378–396. doi:10.1021/cm102117c
Return to citation in text: [1] -
Zhao, K.-Q.; Chen, C.; Monobe, H.; Hu, P.; Wang, B.-Q.; Shimizu, Y. Chem. Commun. 2011, 47, 6290–6292. doi:10.1039/c1cc10299k
Return to citation in text: [1] -
Ni, H.-L.; Monobe, H.; Hu, P.; Wang, B.-Q.; Shimizu, Y.; Zhao, K.-Q. Liq. Cryst. 2013, 40, 411–420. doi:10.1080/02678292.2012.755224
Return to citation in text: [1] -
Monobe, H.; Chen, C.; Zhao, K.-Q.; Hu, P.; Miyake, Y.; Fujii, A.; Ozaki, M.; Shimizu, Y. Mol. Cryst. Liq. Cryst. 2011, 545, 149–155. doi:10.1080/15421406.2011.568891
Return to citation in text: [1] -
Seguy, I.; Jolinat, P.; Destruel, P.; Farenc, J.; Mamy, R.; Bock, H.; Ip, J.; Nguyen, T. P. J. Appl. Phys. 2001, 89, 5442. doi:10.1063/1.1365059
Return to citation in text: [1] -
Hassheider, T.; Benning, S. A.; Kitzerow, H.-S.; Achard, M.-F.; Bock, H. Angew. Chem., Int. Ed. 2001, 40, 2060–2063. doi:10.1002/1521-3773(20010601)40:11<2060::AID-ANIE2060>3.3.CO;2-8
Return to citation in text: [1] -
Freudenmann, R.; Behnisch, B.; Hanack, M. J. Mater. Chem. 2001, 11, 1618–1624. doi:10.1039/b100083g
Return to citation in text: [1] -
Diring, S.; Camerel, F.; Donnio, B.; Dintzer, T.; Toffanin, S.; Capelli, R.; Muccini, M.; Ziessel, R. J. Am. Chem. Soc. 2009, 131, 18177–18185. doi:10.1021/ja908061q
Return to citation in text: [1] -
Mei, J.; Diao, Y.; Appleton, A. L.; Fang, L.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 6724–6746. doi:10.1021/ja400881n
Return to citation in text: [1] -
Schmidt-Mende, L.; Fechtenkötter, A.; Müllen, K.; Moons, E.; Friend, R. H.; MacKenzie, J. D. Science 2001, 293, 1119–1122. doi:10.1126/science.293.5532.1119
Return to citation in text: [1] -
Hayashi, H.; Nihashi, W.; Umeyama, T.; Matano, Y.; Seki, S.; Shimizu, Y.; Imahori, H. J. Am. Chem. Soc. 2011, 133, 10736–10739. doi:10.1021/ja203822q
Return to citation in text: [1] -
Geerts, Y. H.; Debever, O.; Amoto, C.; Sergeyev, S. Beilstein J. Org. Chem. 2009, 5, No. 49. doi:10.3762/bjoc.5.49
Return to citation in text: [1] -
Kumar, S. Liq. Cryst. 2005, 32, 1089–1113. doi:10.1080/02678290500117415
Return to citation in text: [1] -
Imrie, C. T.; Henderson, P. A. Chem. Soc. Rev. 2007, 36, 2096–2124. doi:10.1039/b714102e
Return to citation in text: [1] -
Imrie, C. T.; Henderson, P. A.; Yeap, G.-Y. Liq. Cryst. 2009, 36, 755–777. doi:10.1080/02678290903157455
Return to citation in text: [1] -
Möller, M.; Tsukruk, V.; Wendorff, J. H.; Bengs, H.; Ringsdorf, H. Liq. Cryst. 1992, 12, 17–36. doi:10.1080/02678299208029035
Return to citation in text: [1] -
Maliszewskyj, N. C.; Heiney, P. A.; Josefowicz, J. Y.; Plesnivy, T.; Ringsdorf, H.; Schuhmacher, P. Langmuir 1995, 11, 1666–1674. doi:10.1021/la00005a040
Return to citation in text: [1] -
Tsukruk, V. V.; Bengs, H.; Ringsdorf, H. Langmuir 1996, 12, 754–757. doi:10.1021/la9506133
Return to citation in text: [1] -
Kumar, S.; Schuhmacher, P.; Hendersen, P.; Rego, J.; Ringsdorf, H. Mol. Cryst. Liq. Cryst. 1996, 228, 211–222. doi:10.1080/10587259608034598
Return to citation in text: [1] -
Mahlstedt, S.; Janietz, D.; Stracke, A.; Wendorff, J. H. Chem. Commun. 2000, 15–16. doi:10.1039/a907770g
Return to citation in text: [1] -
Kaller, M.; Staffeld, P.; Haug, R.; Frey, W.; Giesselmann, F.; Laschat, S. Liq. Cryst. 2011, 38, 531–553. doi:10.1080/02678292.2011.558215
Return to citation in text: [1] -
Kaller, M.; Beardsworth, S. J.; Staffeld, P.; Tussetschläger, S.; Gießelmann, F.; Laschat, S. Liq. Cryst. 2012, 39, 607–618. doi:10.1080/02678292.2012.668720
Return to citation in text: [1] -
Gupta, S. K.; Raghunathan, V. A.; Lakshminarayanan, V.; Kumar, S. J. Phys. Chem. B 2009, 113, 12887–12895. doi:10.1021/jp9042254
Return to citation in text: [1] -
Kong, X.; He, Z.; Zhang, Y.; Mu, L.; Liang, C.; Chen, B.; Jing, X.; Cammidge, A. N. Org. Lett. 2011, 13, 764–767. doi:10.1021/ol103018v
Return to citation in text: [1] -
Zelcer, A.; Donnio, B.; Bourgogne, C.; Cukiernik, F. D.; Guillon, D. Chem. Mater. 2007, 19, 1992–2006. doi:10.1021/cm062949b
Return to citation in text: [1] -
Miao, J.; Zhu, L. Chem. Mater. 2010, 22, 197–206. doi:10.1021/cm902731u
Return to citation in text: [1] -
Miao, J.; Zhu, L. J. Phys. Chem. B 2010, 114, 1879–1887. doi:10.1021/jp910053e
Return to citation in text: [1] -
Boden, N.; Bushby, R. J.; Cammidge, A. N.; El-Mansoury, A.; Martin, P. S.; Lu, Z. B. J. Mater. Chem. 1999, 9, 1391–1402. doi:10.1039/a810045d
Return to citation in text: [1] -
El-Mansoury, A.; Bushby, R. J.; Karodia, N. Liq. Cryst. 2012, 39, 1222–1230. doi:10.1080/02678292.2012.707691
Return to citation in text: [1] -
Yang, F.; Xie, J.; Guo, H.; Xu, B.; Li, C. Liq. Cryst. 2012, 39, 1368–1374. doi:10.1080/02678292.2012.717112
Return to citation in text: [1] -
Gupta, S. K.; Kumar, S. Liq. Cryst. 2012, 39, 1443–1449. doi:10.1080/02678292.2012.720289
Return to citation in text: [1] -
Kranig, W.; Hüser, B.; Spiess, H. W.; Kreuder, W.; Ringsdorf, H.; Zimmermann, H. Adv. Mater. 1990, 2, 36–40. doi:10.1002/adma.19900020107
Return to citation in text: [1] -
Paraschiv, I.; Giesbers, M.; van Lagen, B.; Grozema, F. C.; Abellon, R. D.; Siebbeles, L. D. A.; Marcelis, A. T. M.; Zuilhof, H.; Sudhölter, E. J. R. Chem. Mater. 2006, 18, 968–974. doi:10.1021/cm052221f
Return to citation in text: [1] -
Paraschiv, I.; de Lange, K.; Giesbers, M.; van Lagen, B.; Grozema, F. C.; Abellon, R. D.; Siebbeles, L. D. A.; Sudhölter, E. J. R.; Zuilhof, H.; Marcelis, A. T. M. J. Mater. Chem. 2008, 18, 5475–5481. doi:10.1039/b805283b
Return to citation in text: [1] -
Boden, N.; Bushby, R. J.; Cammidge, A. N.; Martin, P. S. J. Mater. Chem. 1995, 5, 1857–1860. doi:10.1039/jm9950501857
Return to citation in text: [1] -
Kumar, S.; Manickam, M. Liq. Cryst. 1999, 26, 939–941. doi:10.1080/026782999204642
Return to citation in text: [1] -
Li, J.; He, Z.; Gopee, H.; Cammidge, A. N. Org. Lett. 2010, 12, 472–475. doi:10.1021/ol902637z
Return to citation in text: [1] -
Zhang, X.-M.; Wang, H.-F.; Wang, S.; Shen, Y.-T.; Yang, Y.-L.; Deng, K.; Zhao, K.-Q.; Zeng, Q.-D.; Wang, C. J. Phys. Chem. C 2013, 117, 307–312. doi:10.1021/jp3095616
Return to citation in text: [1] [2] [3] -
Bai, Y.-F.; Bao, L.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. Liq. Cryst. 2013, 40, 97–105. doi:10.1080/02678292.2012.733034
Return to citation in text: [1] [2] [3] -
Bai, Y.-F.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Redshaw, C. Curr. Org. Chem. 2013, 17, 871–885. doi:10.2174/1385272811317080012
Return to citation in text: [1] [2] [3] -
Yu, W.-H.; Chen, C.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. RSC Adv. 2013, 3, 14099–14105. doi:10.1039/c3ra41874j
Return to citation in text: [1] [2] [3] -
Yu, W.-H.; Nie, S.-C.; Bai, Y.-F.; Jing, Y.; Wang, B.-Q.; Zhao, K.-Q. Sci. China: Chem. 2010, 53, 1134–1141. doi:10.1007/s11426-010-0140-x
Return to citation in text: [1] [2] [3] -
Seyler, H.; Purushothaman, B.; Jones, D. J.; Holmes, A. B.; Wong, W. W. H. Pure Appl. Chem. 2012, 84, 1047–1067. doi:10.1351/PAC-CON-11-09-24
Return to citation in text: [1] [2] -
Xing, C.; Lam, J. W. Y.; Zhao, K.; Tang, B. Z. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2960–2974. doi:10.1002/pola.22631
Return to citation in text: [1] [2] [3] [4] [5] -
Bédard, A.-C.; Collins, S. K. J. Am. Chem. Soc. 2011, 133, 19976–19981. doi:10.1021/ja208902t
Return to citation in text: [1] [2] -
Kabalka, G. W.; Varma, M.; Varma, R. S.; Srivastava, P. C.; Knapp, F. F., Jr. J. Org. Chem. 1986, 51, 2386–2388. doi:10.1021/jo00362a044
Return to citation in text: [1] [2] -
Goswami, A.; Ito, T.; Okamoto, S. Adv. Synth. Catal. 2007, 349, 2368–2374. doi:10.1002/adsc.200700188
Return to citation in text: [1] -
Hilt, G.; Vogler, T.; Hess, W.; Galbiati, F. Chem. Commun. 2005, 11, 1474–1475. doi:10.1039/b417832g
Return to citation in text: [1] -
Zhao, K.-Q.; Zhou, H.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Shimizu, Y. Sci. China: Chem. 2011, 54, 1576–1583. doi:10.1007/s11426-011-4325-8
Return to citation in text: [1] [2] [3] -
Rego, J. A.; Kumar, S.; Ringsdorf, H. Chem. Mater. 1996, 8, 1402–1409. doi:10.1021/cm950582x
Return to citation in text: [1] -
Möller, M.; Wendorff, J. H.; Werth, M.; Spiess, H. W. J. Non-Cryst. Solids 1994, 170, 295–299. doi:10.1016/0022-3093(94)90059-0
Return to citation in text: [1] -
Glüsen, B.; Heitz, W.; Kettner, A.; Wendorff, J. H. Liq. Cryst. 1996, 20, 627–633. doi:10.1080/02678299608031152
Return to citation in text: [1] -
Glüsen, B.; Kettner, A.; Wendorff, J. H. Mol. Cryst. Liq. Cryst. 1997, 303, 115–120. doi:10.1080/10587259708039414
Return to citation in text: [1] -
Glüsen, B.; Kettner, A.; Kopitzke, J.; Wendorff, J. H. J. Non-Cryst. Solids 1998, 241, 113–120. doi:10.1016/S0022-3093(98)00767-4
Return to citation in text: [1] -
Kettner, A.; Wendorff, J. H. Liq. Cryst. 1999, 26, 483–487. doi:10.1080/026782999204912
Return to citation in text: [1] -
Zimmermann, S.; Wendorff, J. H.; Weder, C. Chem. Mater. 2002, 14, 2218–2223. doi:10.1021/cm010932h
Return to citation in text: [1]
| 59. | Möller, M.; Wendorff, J. H.; Werth, M.; Spiess, H. W. J. Non-Cryst. Solids 1994, 170, 295–299. doi:10.1016/0022-3093(94)90059-0 |
| 60. | Glüsen, B.; Heitz, W.; Kettner, A.; Wendorff, J. H. Liq. Cryst. 1996, 20, 627–633. doi:10.1080/02678299608031152 |
| 61. | Glüsen, B.; Kettner, A.; Wendorff, J. H. Mol. Cryst. Liq. Cryst. 1997, 303, 115–120. doi:10.1080/10587259708039414 |
| 62. | Glüsen, B.; Kettner, A.; Kopitzke, J.; Wendorff, J. H. J. Non-Cryst. Solids 1998, 241, 113–120. doi:10.1016/S0022-3093(98)00767-4 |
| 63. | Kettner, A.; Wendorff, J. H. Liq. Cryst. 1999, 26, 483–487. doi:10.1080/026782999204912 |
| 64. | Zimmermann, S.; Wendorff, J. H.; Weder, C. Chem. Mater. 2002, 14, 2218–2223. doi:10.1021/cm010932h |
| 57. | Zhao, K.-Q.; Zhou, H.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Shimizu, Y. Sci. China: Chem. 2011, 54, 1576–1583. doi:10.1007/s11426-011-4325-8 |
| 58. | Rego, J. A.; Kumar, S.; Ringsdorf, H. Chem. Mater. 1996, 8, 1402–1409. doi:10.1021/cm950582x |
| 1. | Bushby, R. J.; Kawata, K. Liq. Cryst. 2011, 38, 1415–1426. doi:10.1080/02678292.2011.603262 |
| 2. | Kawata, K. Chem. Rec. 2002, 2, 59–80. doi:10.1002/tcr.10015 |
| 18. | Schmidt-Mende, L.; Fechtenkötter, A.; Müllen, K.; Moons, E.; Friend, R. H.; MacKenzie, J. D. Science 2001, 293, 1119–1122. doi:10.1126/science.293.5532.1119 |
| 19. | Hayashi, H.; Nihashi, W.; Umeyama, T.; Matano, Y.; Seki, S.; Shimizu, Y.; Imahori, H. J. Am. Chem. Soc. 2011, 133, 10736–10739. doi:10.1021/ja203822q |
| 20. | Geerts, Y. H.; Debever, O.; Amoto, C.; Sergeyev, S. Beilstein J. Org. Chem. 2009, 5, No. 49. doi:10.3762/bjoc.5.49 |
| 57. | Zhao, K.-Q.; Zhou, H.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Shimizu, Y. Sci. China: Chem. 2011, 54, 1576–1583. doi:10.1007/s11426-011-4325-8 |
| 5. | Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c |
| 6. | Shimizu, Y.; Oikawa, K.; Nakayama, K.; Guillon, D. J. Mater. Chem. 2007, 17, 4223–4229. doi:10.1039/b705534j |
| 7. | Funahashi, M. Polym. J. 2009, 41, 459–469. doi:10.1295/polymj.PJ2008324 |
| 8. | O’Neill, M.; Kelly, S. M. Adv. Mater. 2011, 23, 566–584. doi:10.1002/adma.201002884 |
| 16. | Diring, S.; Camerel, F.; Donnio, B.; Dintzer, T.; Toffanin, S.; Capelli, R.; Muccini, M.; Ziessel, R. J. Am. Chem. Soc. 2009, 131, 18177–18185. doi:10.1021/ja908061q |
| 17. | Mei, J.; Diao, Y.; Appleton, A. L.; Fang, L.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 6724–6746. doi:10.1021/ja400881n |
| 52. | Xing, C.; Lam, J. W. Y.; Zhao, K.; Tang, B. Z. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2960–2974. doi:10.1002/pola.22631 |
| 13. | Seguy, I.; Jolinat, P.; Destruel, P.; Farenc, J.; Mamy, R.; Bock, H.; Ip, J.; Nguyen, T. P. J. Appl. Phys. 2001, 89, 5442. doi:10.1063/1.1365059 |
| 14. | Hassheider, T.; Benning, S. A.; Kitzerow, H.-S.; Achard, M.-F.; Bock, H. Angew. Chem., Int. Ed. 2001, 40, 2060–2063. doi:10.1002/1521-3773(20010601)40:11<2060::AID-ANIE2060>3.3.CO;2-8 |
| 15. | Freudenmann, R.; Behnisch, B.; Hanack, M. J. Mater. Chem. 2001, 11, 1618–1624. doi:10.1039/b100083g |
| 55. | Goswami, A.; Ito, T.; Okamoto, S. Adv. Synth. Catal. 2007, 349, 2368–2374. doi:10.1002/adsc.200700188 |
| 56. | Hilt, G.; Vogler, T.; Hess, W.; Galbiati, F. Chem. Commun. 2005, 11, 1474–1475. doi:10.1039/b417832g |
| 3. | Adam, D.; Schuhmacher, P.; Simmerer, J.; Häussling, L.; Siemensmeyer, K.; Etzbachi, K. H.; Ringsdorf, H.; Haarer, D. Nature 1994, 371, 141–143. doi:10.1038/371141a0 |
| 4. | Laschat, S.; Baro, A.; Steinke, N.; Giesselmann, F.; Hägele, C.; Scalia, G.; Judele, R.; Kapatsina, E.; Sauer, S.; Schreivogel, A.; Tosoni, M. Angew. Chem., Int. Ed. 2007, 46, 4832–4887. doi:10.1002/anie.200604203 |
| 5. | Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c |
| 6. | Shimizu, Y.; Oikawa, K.; Nakayama, K.; Guillon, D. J. Mater. Chem. 2007, 17, 4223–4229. doi:10.1039/b705534j |
| 7. | Funahashi, M. Polym. J. 2009, 41, 459–469. doi:10.1295/polymj.PJ2008324 |
| 8. | O’Neill, M.; Kelly, S. M. Adv. Mater. 2011, 23, 566–584. doi:10.1002/adma.201002884 |
| 9. | Kaafarani, B. R. Chem. Mater. 2011, 23, 378–396. doi:10.1021/cm102117c |
| 10. | Zhao, K.-Q.; Chen, C.; Monobe, H.; Hu, P.; Wang, B.-Q.; Shimizu, Y. Chem. Commun. 2011, 47, 6290–6292. doi:10.1039/c1cc10299k |
| 11. | Ni, H.-L.; Monobe, H.; Hu, P.; Wang, B.-Q.; Shimizu, Y.; Zhao, K.-Q. Liq. Cryst. 2013, 40, 411–420. doi:10.1080/02678292.2012.755224 |
| 12. | Monobe, H.; Chen, C.; Zhao, K.-Q.; Hu, P.; Miyake, Y.; Fujii, A.; Ozaki, M.; Shimizu, Y. Mol. Cryst. Liq. Cryst. 2011, 545, 149–155. doi:10.1080/15421406.2011.568891 |
| 52. | Xing, C.; Lam, J. W. Y.; Zhao, K.; Tang, B. Z. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2960–2974. doi:10.1002/pola.22631 |
| 51. | Seyler, H.; Purushothaman, B.; Jones, D. J.; Holmes, A. B.; Wong, W. W. H. Pure Appl. Chem. 2012, 84, 1047–1067. doi:10.1351/PAC-CON-11-09-24 |
| 52. | Xing, C.; Lam, J. W. Y.; Zhao, K.; Tang, B. Z. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2960–2974. doi:10.1002/pola.22631 |
| 52. | Xing, C.; Lam, J. W. Y.; Zhao, K.; Tang, B. Z. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2960–2974. doi:10.1002/pola.22631 |
| 46. | Zhang, X.-M.; Wang, H.-F.; Wang, S.; Shen, Y.-T.; Yang, Y.-L.; Deng, K.; Zhao, K.-Q.; Zeng, Q.-D.; Wang, C. J. Phys. Chem. C 2013, 117, 307–312. doi:10.1021/jp3095616 |
| 47. | Bai, Y.-F.; Bao, L.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. Liq. Cryst. 2013, 40, 97–105. doi:10.1080/02678292.2012.733034 |
| 48. | Bai, Y.-F.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Redshaw, C. Curr. Org. Chem. 2013, 17, 871–885. doi:10.2174/1385272811317080012 |
| 49. | Yu, W.-H.; Chen, C.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. RSC Adv. 2013, 3, 14099–14105. doi:10.1039/c3ra41874j |
| 50. | Yu, W.-H.; Nie, S.-C.; Bai, Y.-F.; Jing, Y.; Wang, B.-Q.; Zhao, K.-Q. Sci. China: Chem. 2010, 53, 1134–1141. doi:10.1007/s11426-010-0140-x |
| 53. | Bédard, A.-C.; Collins, S. K. J. Am. Chem. Soc. 2011, 133, 19976–19981. doi:10.1021/ja208902t |
| 54. | Kabalka, G. W.; Varma, M.; Varma, R. S.; Srivastava, P. C.; Knapp, F. F., Jr. J. Org. Chem. 1986, 51, 2386–2388. doi:10.1021/jo00362a044 |
| 57. | Zhao, K.-Q.; Zhou, H.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Shimizu, Y. Sci. China: Chem. 2011, 54, 1576–1583. doi:10.1007/s11426-011-4325-8 |
| 46. | Zhang, X.-M.; Wang, H.-F.; Wang, S.; Shen, Y.-T.; Yang, Y.-L.; Deng, K.; Zhao, K.-Q.; Zeng, Q.-D.; Wang, C. J. Phys. Chem. C 2013, 117, 307–312. doi:10.1021/jp3095616 |
| 47. | Bai, Y.-F.; Bao, L.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. Liq. Cryst. 2013, 40, 97–105. doi:10.1080/02678292.2012.733034 |
| 48. | Bai, Y.-F.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Redshaw, C. Curr. Org. Chem. 2013, 17, 871–885. doi:10.2174/1385272811317080012 |
| 49. | Yu, W.-H.; Chen, C.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. RSC Adv. 2013, 3, 14099–14105. doi:10.1039/c3ra41874j |
| 50. | Yu, W.-H.; Nie, S.-C.; Bai, Y.-F.; Jing, Y.; Wang, B.-Q.; Zhao, K.-Q. Sci. China: Chem. 2010, 53, 1134–1141. doi:10.1007/s11426-010-0140-x |
| 53. | Bédard, A.-C.; Collins, S. K. J. Am. Chem. Soc. 2011, 133, 19976–19981. doi:10.1021/ja208902t |
| 54. | Kabalka, G. W.; Varma, M.; Varma, R. S.; Srivastava, P. C.; Knapp, F. F., Jr. J. Org. Chem. 1986, 51, 2386–2388. doi:10.1021/jo00362a044 |
| 21. | Kumar, S. Liq. Cryst. 2005, 32, 1089–1113. doi:10.1080/02678290500117415 |
| 22. | Imrie, C. T.; Henderson, P. A. Chem. Soc. Rev. 2007, 36, 2096–2124. doi:10.1039/b714102e |
| 23. | Imrie, C. T.; Henderson, P. A.; Yeap, G.-Y. Liq. Cryst. 2009, 36, 755–777. doi:10.1080/02678290903157455 |
| 24. | Möller, M.; Tsukruk, V.; Wendorff, J. H.; Bengs, H.; Ringsdorf, H. Liq. Cryst. 1992, 12, 17–36. doi:10.1080/02678299208029035 |
| 25. | Maliszewskyj, N. C.; Heiney, P. A.; Josefowicz, J. Y.; Plesnivy, T.; Ringsdorf, H.; Schuhmacher, P. Langmuir 1995, 11, 1666–1674. doi:10.1021/la00005a040 |
| 26. | Tsukruk, V. V.; Bengs, H.; Ringsdorf, H. Langmuir 1996, 12, 754–757. doi:10.1021/la9506133 |
| 27. | Kumar, S.; Schuhmacher, P.; Hendersen, P.; Rego, J.; Ringsdorf, H. Mol. Cryst. Liq. Cryst. 1996, 228, 211–222. doi:10.1080/10587259608034598 |
| 28. | Mahlstedt, S.; Janietz, D.; Stracke, A.; Wendorff, J. H. Chem. Commun. 2000, 15–16. doi:10.1039/a907770g |
| 29. | Kaller, M.; Staffeld, P.; Haug, R.; Frey, W.; Giesselmann, F.; Laschat, S. Liq. Cryst. 2011, 38, 531–553. doi:10.1080/02678292.2011.558215 |
| 30. | Kaller, M.; Beardsworth, S. J.; Staffeld, P.; Tussetschläger, S.; Gießelmann, F.; Laschat, S. Liq. Cryst. 2012, 39, 607–618. doi:10.1080/02678292.2012.668720 |
| 31. | Gupta, S. K.; Raghunathan, V. A.; Lakshminarayanan, V.; Kumar, S. J. Phys. Chem. B 2009, 113, 12887–12895. doi:10.1021/jp9042254 |
| 32. | Kong, X.; He, Z.; Zhang, Y.; Mu, L.; Liang, C.; Chen, B.; Jing, X.; Cammidge, A. N. Org. Lett. 2011, 13, 764–767. doi:10.1021/ol103018v |
| 33. | Zelcer, A.; Donnio, B.; Bourgogne, C.; Cukiernik, F. D.; Guillon, D. Chem. Mater. 2007, 19, 1992–2006. doi:10.1021/cm062949b |
| 34. | Miao, J.; Zhu, L. Chem. Mater. 2010, 22, 197–206. doi:10.1021/cm902731u |
| 35. | Miao, J.; Zhu, L. J. Phys. Chem. B 2010, 114, 1879–1887. doi:10.1021/jp910053e |
| 36. | Boden, N.; Bushby, R. J.; Cammidge, A. N.; El-Mansoury, A.; Martin, P. S.; Lu, Z. B. J. Mater. Chem. 1999, 9, 1391–1402. doi:10.1039/a810045d |
| 37. | El-Mansoury, A.; Bushby, R. J.; Karodia, N. Liq. Cryst. 2012, 39, 1222–1230. doi:10.1080/02678292.2012.707691 |
| 38. | Yang, F.; Xie, J.; Guo, H.; Xu, B.; Li, C. Liq. Cryst. 2012, 39, 1368–1374. doi:10.1080/02678292.2012.717112 |
| 39. | Gupta, S. K.; Kumar, S. Liq. Cryst. 2012, 39, 1443–1449. doi:10.1080/02678292.2012.720289 |
| 40. | Kranig, W.; Hüser, B.; Spiess, H. W.; Kreuder, W.; Ringsdorf, H.; Zimmermann, H. Adv. Mater. 1990, 2, 36–40. doi:10.1002/adma.19900020107 |
| 41. | Paraschiv, I.; Giesbers, M.; van Lagen, B.; Grozema, F. C.; Abellon, R. D.; Siebbeles, L. D. A.; Marcelis, A. T. M.; Zuilhof, H.; Sudhölter, E. J. R. Chem. Mater. 2006, 18, 968–974. doi:10.1021/cm052221f |
| 42. | Paraschiv, I.; de Lange, K.; Giesbers, M.; van Lagen, B.; Grozema, F. C.; Abellon, R. D.; Siebbeles, L. D. A.; Sudhölter, E. J. R.; Zuilhof, H.; Marcelis, A. T. M. J. Mater. Chem. 2008, 18, 5475–5481. doi:10.1039/b805283b |
| 43. | Boden, N.; Bushby, R. J.; Cammidge, A. N.; Martin, P. S. J. Mater. Chem. 1995, 5, 1857–1860. doi:10.1039/jm9950501857 |
| 44. | Kumar, S.; Manickam, M. Liq. Cryst. 1999, 26, 939–941. doi:10.1080/026782999204642 |
| 45. | Li, J.; He, Z.; Gopee, H.; Cammidge, A. N. Org. Lett. 2010, 12, 472–475. doi:10.1021/ol902637z |
| 46. | Zhang, X.-M.; Wang, H.-F.; Wang, S.; Shen, Y.-T.; Yang, Y.-L.; Deng, K.; Zhao, K.-Q.; Zeng, Q.-D.; Wang, C. J. Phys. Chem. C 2013, 117, 307–312. doi:10.1021/jp3095616 |
| 47. | Bai, Y.-F.; Bao, L.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. Liq. Cryst. 2013, 40, 97–105. doi:10.1080/02678292.2012.733034 |
| 48. | Bai, Y.-F.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Redshaw, C. Curr. Org. Chem. 2013, 17, 871–885. doi:10.2174/1385272811317080012 |
| 49. | Yu, W.-H.; Chen, C.; Hu, P.; Wang, B.-Q.; Redshaw, C.; Zhao, K.-Q. RSC Adv. 2013, 3, 14099–14105. doi:10.1039/c3ra41874j |
| 50. | Yu, W.-H.; Nie, S.-C.; Bai, Y.-F.; Jing, Y.; Wang, B.-Q.; Zhao, K.-Q. Sci. China: Chem. 2010, 53, 1134–1141. doi:10.1007/s11426-010-0140-x |
| 51. | Seyler, H.; Purushothaman, B.; Jones, D. J.; Holmes, A. B.; Wong, W. W. H. Pure Appl. Chem. 2012, 84, 1047–1067. doi:10.1351/PAC-CON-11-09-24 |
| 52. | Xing, C.; Lam, J. W. Y.; Zhao, K.; Tang, B. Z. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2960–2974. doi:10.1002/pola.22631 |
© 2013 Han et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)