Abstract
The cis-dibromination of unsaturated bicyclic bridgehead sultams 5a and 5b, and experiments designed to understand the cis-stereochemical outcome of these reactions, are described. In the case of 5b, a novel solvent dependent carbocation rearrangement occurs with the formation of 18b. cis-Dibromides 13a and 13b undergo regioselective dehydrobromination, and the participation of the resultant vinyl bromide 24a in lithiation and Pd-coupling chemistry is described. In the case of the latter, hydrogenation of the styryl products afforded a single diastereoisomer. These compounds were then studied under dissolved metal reduction conditions, in which the cleavage of both N–S and C–S bonds takes place to afford cis-2,4-diaryl-substituted pyrrolidines 35–37.

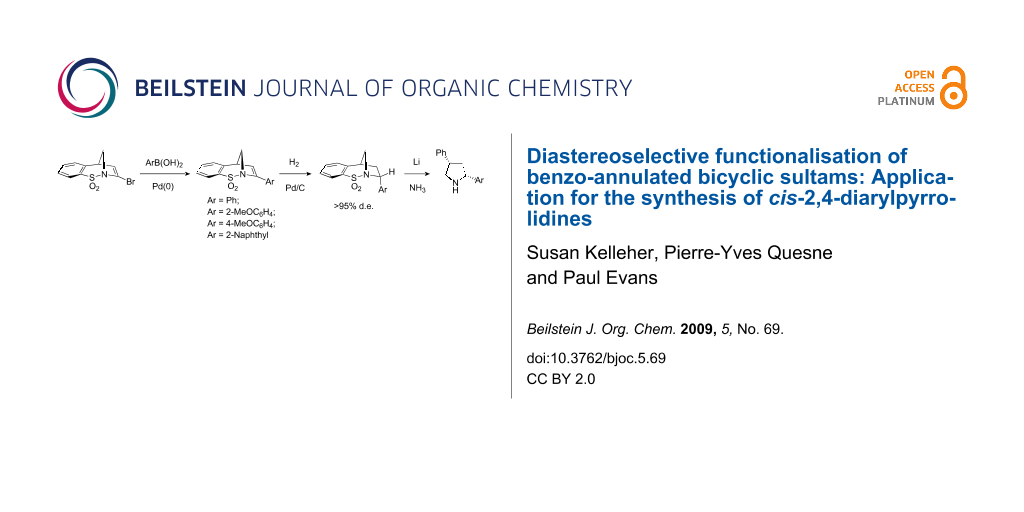
Graphical Abstract
Introduction
Substituted pyrrolidine ring systems represent a common structural motif in a range of biologically active compounds, including pharmaceutical agents and natural products. In relation to these general targets, we have recently developed a method that enables the construction of aryl-substituted pyrrolidines, featuring the double reduction of cyclic aromatic sulfonamides [1-4]. As illustrated in Scheme 1, a Heck (Heck–Mizoroki) cyclisation [5-9] was employed to form the cyclic sulfonamide. Subsequently, it was shown that high stereofacial bias was achieved on hydrogenation, generating 2 as a single diastereoisomer. Treatment under dissolved metal reduction conditions afforded the cis-disubstituted pyrrolidine. In this sequence, the sulfonyl moiety not only serves as an amino protecting group but also facilitates the diastereoselective intramolecular carbon–carbon bond formation. In this present study we demonstrate that this general concept may be extended enabling the diastereoselective preparation of 2,4-diaryl-substituted pyrrolidines from more readily available, albeit racemic, substrates.
![[1860-5397-5-69-i1]](/bjoc/content/inline/1860-5397-5-69-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The diastereoselective intramolecular Heck-hydrogenation and double reduction sequence as a means of accessing cis-disubstituted pyrrolidine 3.
Scheme 1: The diastereoselective intramolecular Heck-hydrogenation and double reduction sequence as a means o...
Results and Discussion
Bicyclic aromatic cyclic sulfonamides 5a and 5b were formed according to the 3-step sequence previously described [1-3,10]. Originally, inclusion of triphenylphosphine was employed, however, optimisation of this reaction with the exclusion of PPh3 gave products 5a and 5b in 82 and 65% yields, respectively, which are comparable with the phosphine-based method (5a: 84% and 5b: 76% with PPh3) [11]. Standard alkenyl hydrogenation (not shown) then gave the saturated bicycles 6a and 6b which were used as substrates for the N–S and C–S bond cleavage forming arylsubstituted pyrrolidines [1-3]. In relation to this sequence attempts to achieve a reductive Heck cyclisation employing ammonium formate [12], gave only the product of bromine-hydrogen exchange 4c (where X = H). Therefore, a one-pot method was developed based on recent reports which demonstrate that the residual palladium catalyst in Heck reactions may effectively mediate the addition of hydrogen to the newly substituted alkene [13-15]. Thus, following complete Heck cyclisation, as judged by TLC analysis, the reaction was simply stirred under a hydrogen atmosphere in order to afford the corresponding saturated bicycles 6a and 6b in yields of 61 and 52% respectively. In terms of efficiency this one-pot process is competitive compared with those from the original two-step, two-pot process (Scheme 2).
![[1860-5397-5-69-i2]](/bjoc/content/inline/1860-5397-5-69-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of 5a and 5b by an intramolecular Heck cyclisation reaction.
Scheme 2: The synthesis of 5a and 5b by an intramolecular Heck cyclisation reaction.
Functionalisation of the N-sulfonyl enamines 5a and 5b was subsequently studied with the ultimate aim of introducing substituents, in a stereoselective fashion, to the masked pyrrolidine ring. Thus, epoxidation of 5a and 5b (Scheme 3) using m-CPBA proceeded smoothly for both substrates and as hoped, occurred under complete stereocontrol (de >95%). The stereochemical outcome was confirmed by single-crystal X-ray crystallography (Figure 1, and see crystallographic data). The use of in situ generated trifluoromethyl-methyldioxirane was also investigated and in the case of 5a, very efficient formation of 7a was observed [16,17].
![[1860-5397-5-69-i3]](/bjoc/content/inline/1860-5397-5-69-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Diastereoselective epoxidation of 5a and 5b.
Scheme 3: Diastereoselective epoxidation of 5a and 5b.
![[1860-5397-5-69-1]](/bjoc/content/figures/1860-5397-5-69-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray structure of 7a; including a view along the S=O···C–O axis (Diamond representations) [O2···C9: 2.850(2) Å].
Figure 1: X-ray structure of 7a; including a view along the S=O···C–O axis (Diamond representations) [O2···C9...
Unfortunately, however, under a variety of conditions it proved impossible to elaborate these species. For example, reductive ring-opening using triethylsilane, conditions previously reported for N-sulfonylamino-α,β-epoxides [18,19], led only to recovery of starting material and not the hoped for alcohol 8. A similar outcome was observed under both Sakurai-type conditions and treatment with potassium cyanide. It seems probable that these failures reflect a combination of the inability of the N-lone pair to stabilise the developing α-carbocation (the resultant N-sulfonyl iminium ion would be anti-Bredt) and in addition the lone pair is perpendicular to the p-orbital resulting from C–O bond cleavage. Furthermore, it also appears that the aromatic ring, based on the fold of the molecule, represents a steric barrier blocking the approach to the C–O σ* orbital. Since the bicyclic structure was implicated in the lack of epoxide reactivity, double reduction of 7a was then considered. It was hoped that following N–S bond cleavage, the now electron rich amino species would form an imine 9 that would then undergo further reduction to afford 11. However, in the event the only isolable product was 12. Although the yield for this process was low, the isolated product indicated that C11 underwent reduction, presumably prior to N–S bond cleavage.
Based on a recent report from Paquette [20], the dibromination of 5a and 5b was subsequently considered. We felt that bromination of the double bond would lead to further possibilities for subsequent functionalisation. This report indicated that the treatment of 5a with neat bromine gave the cis-1,2-dibromide 13a in quantitative yield. It was also reported that under more standard brominating conditions mixtures of diastereoisomeric 1,2-dibromides were formed. In our hands broadly similar results were encountered using chloroform or dichloromethane as reaction solvents (e.g. Entries 1–3). Furthermore, separation of the different diastereoisomers by column chromatography proved difficult. The unusual stereochemical outcome of this bromination process was carefully studied by a combination of NOE analysis, using the diagnostic 12a-CH2 signal, and X-ray crystallography which helped to substantiate the structures of the minor 1,2-trans-diastereoisomers 14a and 15a (see crystallographic data). Optimum conditions, forming 13a as the major diastereomer, were ultimately found (Scheme 4). Thus, addition of bromine to the compound in toluene at low temperature gave 13a in 75% isolated yield and high diastereoselectivity as observed by 1H NMR spectroscopy of the crude reaction mixture (Entry 4).
![[1860-5397-5-69-i4]](/bjoc/content/inline/1860-5397-5-69-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: cis-Selective dibromination of 5a (NOE indicated by arrows).
Scheme 4: cis-Selective dibromination of 5a (NOE indicated by arrows).
In an attempt to further understand the mechanism of this bromination reaction, a bromo-methanolysis reaction was carried out (Entry 5). Interestingly, regio- and diastereoselective formation of the cis-16a was observed in good yield. When 13a was taken up in methanol no conversion into 16a, via an SN1-type process, was observed. The use of THF as solvent led to the formation of compound 17a, albeit in low yield, in which a molecule of the solvent has been incorporated (Entry 6). For X-ray crystal structure of 13a see Figure 2.
![[1860-5397-5-69-2]](/bjoc/content/figures/1860-5397-5-69-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of 13a (Diamond representation).
Figure 2: X-ray crystal structure of 13a (Diamond representation).
The analogous functionalisation of alkene 5b was then considered. In toluene (Scheme 5, Entry 1), the optimal solvent for the cis-dibromination of 5b, as hoped, selective formation of 13b was observed (>90% by 1H NMR spectroscopy of the crude mixture). It should be noted that whilst the selectivity of this reaction is good, it did prove difficult to perform in a reproducible manner based on the limited solubility of 5b in toluene and competing benzyl bromide formation in some instances.
![[1860-5397-5-69-i5]](/bjoc/content/inline/1860-5397-5-69-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
When the same reaction was conducted in chloroform, the formation of 18b as the major product (42% yield) was observed (Entry 2). The identity of this compound was determined by single crystal X-ray crystallography (Figure 3). Strikingly, none of this rearranged type of product was observed for 5a under identical conditions. The yield for this process was increased to 87% when an excess of Br2 (10 equiv) was employed. The bromo-methanolysis reaction, performed as described above, gave a mixture of compounds 16b and 19b in 58% and 37% yields, respectively (Entry 3). The structure of 19b was also confirmed by X-ray crystallography (see Figure 3 and crystallographic data).
![[1860-5397-5-69-3]](/bjoc/content/figures/1860-5397-5-69-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structures of 18b and 19b (Diamond representations).
Figure 3: X-ray crystal structures of 18b and 19b (Diamond representations).
Based on the results obtained we propose a mechanistic explanation illustrated in Scheme 6 to account for the products formed. It seems reasonable to speculate that the cis-1,2-dibromide formation results from the slow reaction of the bromonium ion 20 with bromide due to the blocking of the lower face by the aromatic ring (route c). Indeed, the epoxide 7a (Figure 1) may be considered a bromonium ion model and inspection of this representation demonstrates the pronounced fold of this structure. Consequently, since the SN2 process is retarded the bromide may then intercept either carbocation 21, or 22 in an SN1 sense, from the less hindered, top face, forming 13a and 13b. Using methanol as the solvent the cis-1,2-difunctionalised compounds 16a and 16b were formed regioselectively (none of the products resulting from methanolysis of 22 were detected). The regiochemical outcome of this process suggests that either the carbocation adjacent to nitrogen is energetically more stable, or that it is more reactive under these conditions. In relation to the N-sulfonyl group and its ability to stabilise an α-carbocation, N-sulfonyl iminium ions have been implicated in many instances [18,19,21]. However, in this particular case, based on the conformation of the rigid bicyclic molecule, this mode of stabilisation is not feasible. An alternative mechanism of stabilisation has been suggested in which one of the sulfonyl oxygen atoms may act as a Lewis base and in doing so stabilise the positive charge [22,23]. Inspection of the X-ray structure obtained for compounds 7a and 7b (not shown) indicates that this stabilisation mechanism may be possible based on the distance between the carbon atom of the epoxide and its closest sulfonyl oxygen atom (see Figure 1).
![[1860-5397-5-69-i6]](/bjoc/content/inline/1860-5397-5-69-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible explanation for the products formed in the dibromination of 5a and 5b.
Scheme 6: Possible explanation for the products formed in the dibromination of 5a and 5b.
In the case of carbocation 22, the benzylic bond appears to be aligned with the empty p-orbital, consequently, when the aromatic ring is substituted with the +M methoxy substituents the results obtained indicate that a Wagner–Meerwein-type rearrangement occurs to generate carbocation 23. Depending on the conditions this species is then intercepted, again diastereoselectively, by bromide or methanol, affording 18b and 19b, respectively. In the case of 5b in chloroform and methanol this carbocation rearrangement takes place faster than any nucleophilic interception of species 21 and 22. (A hydride shift from the bridging CH2 to carbocation 21 was also considered; however, based on the regioselective formation of compound 19b this mechanism was discounted.) The remarkable contrasting behaviour for compound 5b in chloroform and toluene appears to be explained based on the polarity of the respective solvents. Toluene with a low dielectric constant favours formation of 13b whereas the more polar chloroform facilitates the rearrangement chemistry via carbocation 22. This type of terpene-like carbocation rearrangement has been reported for molecules which might be considered related to our [2.2.1]-bicyclic systems and has been termed a molecular somersault [24].
Regioselective elimination of the cis-dibromides 13a and 13b with TBAF, according to Paquette’s procedure [20], gave the vinyl bromides 24a and 24b in good yields, respectively (Scheme 7). Vinyl bromide 24a was then subjected to lithiation with t-BuLi, followed by a CO2 quench. The desired carboxylic acid recovered from this reaction was only formed in low yields possibly due to issues with competing directed ortho-lithiation [25]. Notwithstanding, this acid was transformed into the corresponding methyl ester 25 using a Steglich-type esterification. Alkenyl reduction of the α,β-unsaturated ester under standard conditions gave the product of hydrogenation as an undetermined 3:1 mixture of diastereoisomers. The likely explanation for this mixture of products, based on the high levels of diastereoselectivity observed in the similar examples discussed herein, is that the initially formed product begins to undergo epimerisation in order to place the larger (CO2Me) substituent on the least hindered face. Consequently, the complete epimerisation of this material was investigated using NaOMe (1 M) based on a literature report [26]. Pleasingly, the hoped for process did indeed lead to the formation of one diastereomer of carboxylic acid 26, which was formed from the methyl ester on hydrolysis following work-up (see Figure 4 and crystallographic data). Although the yields described during this sequence are low at this stage, the successful epimerisation observed demonstrates that this approach represents a stereo-complementary method to those described in Scheme 1 and Scheme 8.
![[1860-5397-5-69-i7]](/bjoc/content/inline/1860-5397-5-69-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Lithiation–CO2 quench approach for the synthesis of 26 from vinyl bromide 24a.
Scheme 7: Lithiation–CO2 quench approach for the synthesis of 26 from vinyl bromide 24a.
![[1860-5397-5-69-4]](/bjoc/content/figures/1860-5397-5-69-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray structure of 26 (Diamond representation).
Figure 4: X-ray structure of 26 (Diamond representation).
With the lithiation chemistry described above proving inefficient, palladium chemistry was subsequently considered. The vinyl bromide 24a was reported to participate in Sonagashira alkynylation chemistry; however, yields of the resultant enyne were low [20]. Therefore, we decided to investigate Suzuki–Miyaura cross-coupling reaction as a means of incorporating alternative structural features into our masked pyrrolidine ring. Under standard conditions, the reactivity of a series of commercially available aryl and alkenyl boronic acids with 24a was investigated. Pleasingly, it was found that in the case of the aryl boronic acids, good yields of the resultant styryl adducts 27–30 were observed. It was found that employment of the boronic acids in excess (approximately 5 equiv) in conjunction with base work-up gave the desired products in good yields with no starting material 24a, or the product of bromine–hydrogen exchange 5a. Unfortunately, under identical conditions no cross-coupled products were obtained following several attempts using Z-crotyl boronic acid and vinyl boronic acid.
![[1860-5397-5-69-i8]](/bjoc/content/inline/1860-5397-5-69-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Suzuki–Miyaura cross-coupling of 24a and the diastereoselective hydrogenation of the resultant styrene adducts.
Scheme 8: Suzuki–Miyaura cross-coupling of 24a and the diastereoselective hydrogenation of the resultant styr...
Hydrogenation of the trisubstituted alkene in compounds 27–30 was carried out in DMF due to the poor solubility of these compounds in more standard solvents. These reactions also proved rather slow. Nevertheless, after 72 h under a hydrogen atmosphere in the presence of Pd/C, moderate to good yields of compounds 31–34 were achieved. As predicted, the addition of hydrogen took place in a diastereoselective manner and the adducts were formed as single diastereoisomers. This sense of diastereoselectivity was confirmed by a series of NOE experiments and the single X-ray crystal structure obtained for compound 31 (see Figure 5 and crystallographic data). Poor conversion was observed when the one-pot reductive-palladium method, analogous to that described in Scheme 2, was attempted.
![[1860-5397-5-69-5]](/bjoc/content/figures/1860-5397-5-69-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: X-ray crystal structure of 31 (Diamond representation).
Figure 5: X-ray crystal structure of 31 (Diamond representation).
The double reduction of compounds 31–34 was next considered. Cleavage of both the S–N and S–C bonds would result in the formation of a series of cis-diaryl-substituted pyrrolidine ring containing compounds. Consequently, treatment of each compound under the currently optimal conditions (lithium in liquid ammonia at −78 °C) gave the products of reduction which were converted into the corresponding tosyl derivatives to aid characterisation and purification (Scheme 9).
![[1860-5397-5-69-i9]](/bjoc/content/inline/1860-5397-5-69-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Attempted sulfonamide double reduction of compounds 31–34.
Scheme 9: Attempted sulfonamide double reduction of compounds 31–34.
The successful double reduction of compounds 31–33 was observed in moderate/low yields. In the case of compound 31, a significant amount of the product 39 of benzylic cleavage was also detected. This type of product was not observed for the more electron rich, methoxy-containing substituents, presumably since the Birch-type radical anionic intermediate, which enables the elimination of the amino group, is disfavoured in this instance. In the case of compound 34, trace amount of the hoped for product 38 was detected. However, in this case the major product 40 stems from the partial reduction of the naphthyl ring in addition to cleavage of the sulfonyl tether.
Conclusion
In summary, the work described demonstrates that we were able to utilise the functionalisation of bicyclic sulfonamide 5a featuring a Suzuki coupling and a diastereoselective hydrogenation to construct cis-2,4-diarylpyrrolidines in a diastereoselective manner. The epimerisation reaction which led to the formation of carboxylic acid 26 demonstrates, in principle, how the trans-2,4-substituted series might also be accessible. The cis-diastereoselective dibromination of the bridgehead sultams 5a and 5b was also studied from a mechanistic perspective and related to this an unusual molecular somersault was uncovered, the occurrence of which was dependent on the electronic nature of the aryl motif and of the reaction solvent.
Experimental
Representative experimental procedures are shown for compounds 7a, 13a, 18b, 24a, 27, 31, 35, 39. For full experimental details see accompanying Supporting Information.
(±)-(1S,10R,11R)-Epoxy-8-thia-9-azatricyclo[7.2.1.02,7]dodeca-2(7),3,5-triene-8,8-dioxide (7a): To a solution of alkene 5a (252 mg, 1.22 mmol, 1 equiv) in CH2Cl2 (50 mL) at 0 °C, 70% (w/w) m-CPBA (1.68 g, 6.82 mmol, 5.5 equiv) was added. The reaction mixture was stirred for 72 h during which time room temperature was reached. The reaction was quenched with a saturated Na2SO3 solution (15 mL) which was basified after 0.5 h with a saturated NaHCO3 solution (15 mL). H2O (10 mL) was added and the aqueous layer was then extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were dried over MgSO4. Filtration followed by solvent removal in vacuo afforded the crude product, which was purified by flash column chromatography (c-Hex–EtOAc; 5:1) affording 7a (179 mg, 66%) as a colourless solid. Recrystallisation from EtOAc gave crystals suitable for X-ray crystallographic analysis. mp 164–165 °C (EtOAc); Rf = 0.45 (c-Hex–EtOAc; 1:1); 1H NMR (300 MHz, CDCl3): δ 3.27 (d, J = 4.0 Hz, 1H, 1-CH), 3.35 (dd, J = 4.0, 12.5 Hz, 1H, 1H, 12a-CH2), 3.65 (s, 1H, 11-CH), 3.92 (d, J = 12.5 Hz, 1H, 12b-CH2), 5.03 (s, 1H, 10-CH), 7.19 (d, J = 6.5 Hz, 1H, ArH), 7.40–7.47 (m, 2H, ArH), 7.67–7.80 (m, 1H, ArH); 13C NMR (100 MHz, CDCl3): δ 39.5 (CH), 49.4 (CH2), 57.8 (CH), 65.4 (CH), 127.0 (CH), 127.7 (CH), 130.1 (CH), 132.5 (CH), 135.3 (C), 136.3 (C); υmax (CH2Cl2/cm−1) (KCl) 3067, 3037, 1472, 1381, 1335, 1246, 1205, 1170, 1061, 1007, 935, 916, 855, 811, 792, 767, 750, 715, 609; m/z (ES) required 224.0379 (MH+, 100%); found 224.0381 (−1.1 ppm); Anal. Calcd for C10H9NO3S: C, 53.80; H, 4.06; N, 6.27. Found: C, 53.63; H, 4.04; N, 6.06.
(±)-(1S,10R,11R)-10,11-Dibromo-8-thia-9-azatricyclo[7.2.1.02,7]dodeca-2(7),3,5-triene-8,8-dioxide (13a) [20]: To a solution of alkene 5a (50 mg, 0.24 mmol, 1.2 equiv) in PhMe (10 mL) at −78 °C, Br2 (0.01 mL, 0.20 mmol, 1 equiv) was added. The reaction mixture was stirred for 15 h during which time, room temperature was reached. The reaction was quenched with a saturated solution of Na2SO3 (10 mL) and H2O (10 mL) was added. The resultant aqueous layer was then further extracted with CH2Cl2 (2 × 10 mL) and the combined organic extracts were dried over MgSO4. Filtration followed by solvent removal in vacuo afforded the crude product, which was purified by flash column chromatography (c-Hex–EtOAc; 9:1) affording 13a (55 mg, 75%) as a colourless solid. mp 174–176 °C (EtOAc), lit. 185–188 °C [20]; Rf = 0.6 (c-Hex–EtOAc; 1:1); 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 1H, 1-CH), 4.10 (d, J = 13.0 Hz, 1H, 12a-CH2), 4.30 (d, J = 13.0 Hz, 1H, 12b-CH2), 4.66 (d, J = 6.0 Hz, 1H, 11-CH), 6.47 (d, J = 6.0 Hz, 1H, 10-CH), 7.35 (d, J = 7.0 Hz, 1H, ArH), 7.50–7.60 (m, 2H, ArH), 7.81 (d, J = 7.0 Hz, 1H, ArH); 13C NMR (100 MHz, CDCl3): δ 52.3 (CH), 52.8 (CH2), 57.8 (CH), 67.6 (CH), 126.6 (CH), 126.7 (CH), 130.5 (CH), 133.7 (CH), 134.9 (C), 135.9 (C); υmax (CH2Cl2/cm−1) (NaCl) 3010, 2977, 1446, 1344, 1324, 1300, 1263, 1234, 1206, 1162, 1077; Anal. Calcd for C10H9Br2NO2S: C, 32.70; H, 2.45; N, 3.89; Br, 43.60. Found: C, 32.66; H, 2.31; N, 3.41; Br, 43.27.
(±)-(1R,11R,12S)-11,12-Dibromo-4,5-dimethoxy-8-thia-9-azatricyclo[7.2.1.02,7]dodeca-2(7),3,5-triene-8,8-dioxide (18b): To a solution of alkene 5b (75 mg, 0.28 mmol, 1 equiv) in CHCl3 (10 mL) at −78 °C, Br2 (0.15 mL, 2.80 mmol, 10 equiv) was added. The reaction mixture was stirred for 15 h during which time, room temperature was reached. The reaction was quenched with a saturated solution of Na2SO3 (20 mL) and H2O (20 mL) was added. The aqueous layer was then further extracted with CH2Cl2 (2 × 20 mL) and the combined organic extracts were dried over MgSO4. Filtration followed by solvent removal in vacuo afforded the crude product, which was purified by flash column chromatography (c-Hex–EtOAc; 5:1) affording 18b (104 mg, 87%) as a colourless solid. Recrystallisation from EtOAc gave crystals suitable for X-ray crystallographic analysis. mp 225 °C (EtOAc); Rf = 0.6 (c-Hex–EtOAc; 1:1); 1H NMR (500 MHz, CDCl3): δ 3.86 (s, 1H, 1-CH), 3.91 (s, 3H, CH3), 3.96 (s, 3H, CH3), 4.13 (dd, J = 5.0, 8.0 Hz, 1H, 11-CH), 4.20 (dd, J = 5.0, 15.0 Hz, 1H, 10a-CH2), 4.60 (dd, J = 8.0, 15.0 Hz, 1H, 10b-CH2), 6.32 (s, 1H, 12-CH), 6.70 (s, 1H, ArH), 7.19 (s, 1H, ArH); 13C NMR (125 MHz, CDCl3): δ 44.0 (CH), 56.3 (CH), 56.4 (CH3), 58.5 (CH3), 58.9 (CH), 64.5 (CH2), 107.8 (CH), 108.5 (CH), 126.3 (C), 130.2 (C), 150.4 (C), 153.2 (C); υmax (CH2Cl2/cm−1) (NaCl) 3096, 2975, 1593, 1444, 1326, 1260, 1203, 1164, 1044; Anal. Calcd for C12H13Br2NO4S: C, 33.75; H, 3.07; N, 3.28. Found: C, 33.69; H, 2.99; N, 3.17.
10-Bromo-8-thia-9-azatricyclo[7.2.1.02,7]dodeca-2,4,6,10-tetraene-8,8-dioxide (24a) [20]: Compound 13a (1.04 g, 2.83 mmol, 1 equiv) in THF (15 mL) was treated with a 1 M solution of TBAF in THF (20 mL, 20.00 mmol, 7 equiv) for 15 h. The solvent was removed in vacuo and CH2Cl2 was added (30 mL). The organic layer was washed with a saturated solution of NaHCO3 (30 mL), the aqueous phase further extracted with CH2Cl2 (30 mL) and the combined organic layers were dried over MgSO4. Filtration followed by solvent removal in vacuo afforded the crude product, which was purified by flash column chromatography (c-Hex–EtOAc; 1:1) affording 24a (700 mg, 86%) as a colourless solid. mp 188–192 °C (CH2Cl2); Rf = 0.3 (c-Hex–EtOAc; 1:1); 1H NMR (400 MHz, CDCl3): δ 3.29 (t, J = 4.0 Hz, 1H, CH), 4.37 (dd, J = 4.0, 12.0 Hz, 1H, 12a-CH2), 4.59 (d, J = 12.0 Hz, 1H, 1H, 12b-CH2), 6.68 (d, J = 4.0 Hz, 1H, 11-CH), 7.12 (d, J = 7.5 Hz, 1H, ArH), 7.42 (t, J = 7.5 Hz, 1H, ArH), 7.50 (t, J = 7.5 Hz, 1H, ArH), 7.77 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (100 MHz, CDCl3): δ 43.9 (CH), 65.5 (CH2), 125.0 (C), 125.4 (CH), 127.4 (CH), 130.2 (CH), 132.1 (CH), 134.5 (C), 135.9 (CH), 139.3 (C); υmax (CH2Cl2/cm−1) (NaCl) 3102, 2924, 1591, 1452, 1338, 1168, 1054, 925, 866, 749; m/z (ES) required 285.9549 (MH+(Br79), 100%); found 285.9537 (+4.1 ppm); Anal. Calcd for C10H8BrNO2S: C, 41.96; H, 2.80; N, 4.90; Found: C, 41.97; H, 2.77; N, 4.71.
10-Phenyl-8-thia-9-azatricyclo[7.2.1.02,7]dodeca-2,4,6,10-tetraene-8,8-dioxide (27): Under N2, a mixture of the compound 24a (500 mg, 1.75 mmol, 1 equiv), phenylboronic acid (1.073 g, 8.80 mmol, 5 equiv), Pd(OAc)2 (20 mg, 0.09 mmol, 5 mol %), PPh3 (47 mg, 0.18 mmol, 10 mol %) and Cs2CO3 (1.714 g, 5.26 mmol, 3 equiv) in a mixture of THF:H2O (10:1) (25 mL) was heated to reflux for 15 h. On cooling Et2O (20 mL) and H2O (20 mL) were added and the resultant aqueous layer was further extracted with Et2O (2 × 20 mL) and the combined organic extracts were washed with a 2 M NaOH solution (20 mL) and dried over MgSO4. Filtration followed by solvent removal under reduced pressure gave the crude product, which was purified by flash column chromatography (c-Hex–EtOAc; 5:1) affording 27 (314 mg, 63%) as a colourless solid. mp 120–122 °C (EtOAc); Rf = 0.55 (c-Hex–EtOAc; 1:1); 1H NMR (400 MHz, CDCl3): δ 3.44 (t, J = 4.0 Hz, 1H, CH), 4.27 (dd, J = 4.0, 12.0 Hz, 1H, CH2), 4.70 (d, J = 12.0 Hz, 1H, CH2), 6.82 (d, J = 4.0 Hz, 1H, CH), 7.17 (d, J = 7.5 Hz, 1H, ArH), 7.31–7.39 (m, 3H, ArH), 7.40–7.47 (m, 2H, ArH), 7.69–7.71 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3): δ 43.3 (CH), 64.3 (CH2), 110.0 (C), 125.3 (CH), 126.6 (CH), 127.1 (CH), 127.3 (CH), 128.3 (CH), 129.2 (CH), 129.7 (CH), 131.7 (CH), 134.7 (C), 140.8 (C), 147.0 (C); υmax (CH2Cl2/cm−1) (KCl) 3063, 1965, 1592, 1452, 1327, 1162, 1029, 948; m/z (ES) required 284.0733 (MH+, 100%); found 284.0745 (−4.3 ppm); Anal. Calcd for C16H13NO2S: C, 67.84; H, 4.59; N, 4.95. Found: C, 67.69; H, 4.65; N, 4.84.
(±)-(1S,10R)-10-Phenyl-8-thia-9-azatricyclo[7.2.1.02,7]dodeca-2(7),3,5-triene-8,8-dioxide (31): 10% (w/w) Pd/C (98 mg, 0.09 mmol, 15 mol %) was added to a solution of the alkene 27 (173 mg, 0.61 mmol, 1 equiv) in DMF (20 mL). The mixture was degassed before stirring under a hydrogen atmosphere (1 atm) at room temperature for 72 h. Filtration through Celite® and solvent removal in vacuo gave 32 (77 mg, 44%) as a colourless crystalline solid. mp 131–134 °C (EtOAc); Rf = 0.5 (c-Hex–EtOAc; 1:1); 1H NMR (400 MHz, CDCl3): δ 2.36 (ddd, J = 2.0, 7.5, 13.0 Hz, 1H, CH2), 2.73 (ddd, J = 7.5, 10.0, 13.0 Hz, 1H, CH2), 3.43 (dd, J = 3.5, 7.5 Hz, 1H, CH), 3.56 (dd, J = 3.5, 12.5 Hz, 1H, CH2), 4.53 (dd, J = 2.0, 12.5 Hz, 1H, CH2), 5.05 (dd, J = 7.5, 10.0 Hz, 1H, CH), 7.13–7.19 (m, 2H, ArH), 7.22–7.29 (m, 4H, ArH), 7.36 (dt, J = 1.0, 7.0 Hz, 1H, ArH), 7.48 (dt, J = 1.0, 7.5 Hz, 1H, ArH), 7.66 (d, J = 7.5 Hz, 1H, 1H, ArH); 13C NMR (100 MHz, CDCl3): δ 37.7 (CH2), 40.1 (CH), 59.1 (CH2), 66.4 (CH), 125.8 (CH), 127.0 (CH), 127.9 (CH), 128.2 (CH), 128.3 (CH), 129.8 (CH), 132.6 (CH), 135.1 (C), 137.2 (C), 142.1 (C); υmax (CH2Cl2/cm−1) (KCl) 3042, 2956, 2342, 1954, 1876, 1596, 1452, 1324, 1163, 1087, 975; m/z (ES) required 284.0894 (MH+, 100%); found 284.0902 (−2.7 ppm).
(±)-(2R,4S)-2,4-Diphenyl-1-(toluene-4-sulfonyl)pyrrolidine (35) and N-(2,4-diphenylbutyl)-4-methylbenzenesulfonamide (39): Under N2 at −78 °C liquid NH3 (ca. 100 mL) was treated with lithium wire (10 mg, 1.43 mmol, 5 equiv). This mixture was stirred for 1 h before a solution of 31 (80 mg, 0.28 mmol, 1 equiv) in THF (5 mL) was introduced dropwise. Stirring was continued at −78 °C for 20 min before solid NH4Cl (ca. 5 g) was added. The NH3 was allowed to evaporate and Et2O (25 mL) and H2O (25 mL) were added to the residue. The resultant aqueous layer was further extracted with Et2O (2 × 25 mL) and the combined organic extracts were dried over MgSO4. Filtration, followed by solvent removal under reduced pressure, afforded a colourless oil. At 0 °C the crude material was treated with Et3N (0.06 mL, 0.43 mmol, 1.5 equiv) and TsCl (53 mg, 0.28 mmol, 1 equiv) in CH2Cl2 (10 mL). The mixture was stirred for 3 h during which time, room temperature was reached. CH2Cl2 (20 mL) and H2O (20 mL) were added and the resultant aqueous layer was further extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were dried over MgSO4, which was removed by filtration. The solvent was removed under reduced pressure and purification of the residue by flash column chromatography (c-Hex–EtOAc; 4:1) gave initially 35 (21 mg, 20%) as a colourless oil. Rf = 0.45 (c-Hex–EtOAc; 3:1); 1H NMR (400 MHz, CDCl3): δ 2.05 (ddd, app. dt, J = 10.0, 12.5 Hz, 1H, CH2), 2.43 (s, 3H, CH3), 2.63–2.72 (m, 1H, CH2), 2.90–3.02 (m, 1H, CH), 3.51 (t, J = 11.0 Hz, 1H, CH2), 4.16 (ddd, J = 1.0, 7.0, 11.0 Hz, 1H, CH2), 4.82 (dd, J = 7.0, 10.0 Hz, 1H, CH), 7.12 (d, J = 7.0 Hz, 2H, ArH), 7.20–7.36 (m, 10H, ArH), 7.63 (d, J = 8.0 Hz, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ 21.5 (CH3), 43.7 (CH), 44.4 (CH2), 55.9 (CH2), 64.5 (CH), 126.4 (CH), 127.0 (CH), 127.1 (CH), 127.3 (CH), 127.4 (CH), 128.4 (CH), 128.7 (CH), 129.6 (CH), 135.7 (C), 139.0 (C), 142.5 (C), 143.3 (C); υmax (CH2Cl2/cm−1) 3059, 2926, 1599, 1494, 1432, 1342, 1289, 1254, 1158, 1093, 1027; m/z (ES) required 378.1535 (MH+, 100%); found 378.1528 (+1.1 ppm). Further elution gave 39 (51 mg, 48%) as a colourless oil. Rf = 0.35 (c-Hex–EtOAc; 3:1); 1H NMR (300 MHz, CDCl3): δ 1.77–1.88 (m, 1H, CH2), 1.90–2.00 (m, 1H, CH2), 2.34–2.49 (m, 5H, CH3, CH2), 2.63–2.72 (m, 1H, CH), 3.00 (app. ddt, J = 4.5, 9.0 Hz, 1H, CH2), 3.27 (app. ddt, J = 5.5, 8.0 Hz, 1H, CH2), 4.21 (dd, J = 4.0, 4.5 Hz, 1H, NH), 7.01–7.06 (m, 4H, ArH), 7.13–7.35 (m, 8H, ArH), 7.62 (d, J = 8.0 Hz, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ 21.5 (CH3), 33.1 (CH2), 35.0 (CH2), 45.0 (CH), 48.5 (CH2), 125.9 (CH), 127.0 (CH), 127.3 (CH), 127.8 (CH), 128.3 (CH), 128.35 (CH), 129.0 (CH), 129.6 (CH), 137.0 (C), 141.0 (C), 141.5 (C), 143.3 (C); υmax (CH2Cl2/cm−1) 3360, 2043, 2932, 1599, 1493, 1431, 1327, 1222, 1159, 1087, 810; m/z (ES) required 380.1666 (MH+, 100%); found 380.1684 (−4.8 ppm).
Crystallographic data
Crystal structural data for compound 7a: C10H9NO3S; M = 223.24; orthorhombic, Pbca; a = 8.9434(12) Å, b = 12.1447(16) Å, c = 17.0400(2) Å; U = 1850.8(4) Å3; T = 100(2) K; Z = 8; 15241 reflections measured, 2019 unique (Rint = 0.0514). The final wR2 was 0.1169 (all data). CCDC reference number 742466. Crystal structure data for compound 13a: C10H9NO2SBr2; M = 367.06; monoclinic, P21/c; a = 11.1268(11) Å, b = 8.6567(8) Å, c = 12.5715(12) Å; U = 1129.8(19) Å3; T = 100(2) K; Z = 4; 11016 reflections measured, 2808 unique (Rint = 0.0407). The final wR2 was 0.0671 (all data). CCDC reference number 742467. Crystal structural data for compound 18b: C12H13NO4SBr2; M = 427.11; monoclinic, P21/n; a = 11.462(2) Å, b = 10.4508(18) Å, c = 12.0450(2) Å; U = 1405.2(4) Å3; T = 100(2) K; Z = 4; 11889 reflections measured, 2867 unique (Rint = 0.0378). The final wR2 was 0.0946 (all data). CCDC reference number 742468. Crystal structural data for compound 19b: C13H16NO5SBr; M = 378.24; monoclinic, P21/c; a = 15.9581(19) Å, b = 10.6318(13) Å, c = 17.8100(2) Å; U = 2889.5(6) Å3; T = 100(2) K; Z = 8; 65857 reflections measured, 8789 unique (Rint = 0.0329). The final wR2 was 0.0712 (all data). CCDC reference number 742469. Crystal structural data for compound 26: C11H11NO4S; M = 253.27; monoclinic, P21/n; a = 14.7519(16) Å, b = 9.6485(10) Å, c = 15.2476(16) Å; U = 2163.8(4) Å3; T = 100(2) K; Z = 8; 37265 reflections measured, 4487 unique (Rint = 0.0384). The final wR2 was 0.1158 (all data). CCDC reference number 742471. Crystal structural data for compound 31: C16H15NO2S; M = 285.35; orthorhombic, P212121; a = 9.3949(7) Å, b = 10.7576(8) Å, c = 13.5221(10) Å; U = 1366.63(18) Å3; T = 180(2) K; Z = 4; 16599 reflections measured, 4468 unique (Rint = 0.0157). The final wR2 was 0.0852 (all data). CCDC reference number 742470.
Supporting Information
Supporting information features experimental procedures and spectroscopic analyses for compounds 6a, 6b, 7a, 7b, 13a, 13b, 16a, 16b, 17a, 18b, 19b, 24a, 24b, 25–37, and 39.
| Supporting Information File 1: Diastereoselective functionalisation of benzo-annulated bicyclic sultams: application for the synthesis of cis-2,4-diarylpyrrolidines. | ||
| Format: PDF | Size: 242.8 KB | Download |
References
-
Evans, P.; McCabe, T.; Morgan, B. S.; Reau, S. Org. Lett. 2005, 7, 43–46. doi:10.1021/ol0480123
Return to citation in text: [1] [2] [3] -
Evans, P. J. Org. Chem. 2007, 72, 1830–1833. doi:10.1021/jo062189o
Return to citation in text: [1] [2] [3] -
Kelleher, S.; Muldoon, J.; Müller-Bunz, H.; Evans, P. Tetrahedron Lett. 2007, 48, 4733–4736. doi:10.1016/j.tetlet.2007.05.015
Return to citation in text: [1] [2] [3] -
Zeng, W.; Chemler, S. R. J. Org. Chem. 2008, 73, 6045–6047. doi:10.1021/jo801024h
Return to citation in text: [1] -
Knowles, J. P.; Whiting, A. Org. Biomol. Chem. 2007, 5, 31–44. doi:10.1039/b611547k
(see for recent reviews).
Return to citation in text: [1] -
Zeni, G.; Larock, R. C. Chem. Rev. 2006, 106, 4644–4680. doi:10.1021/cr0683966
(see for recent reviews).
Return to citation in text: [1] -
Gibson, S. E.; Middleton, R. J. Contemp. Org. Synth. 1996, 3, 447–471. doi:10.1039/CO9960300447
(see for recent reviews).
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
(see for recent reviews).
Return to citation in text: [1] -
Link, J. T. Org. React. 2002, 60, 157–534. doi:10.1002/0471264180.or060.02
(see for recent reviews).
Return to citation in text: [1] -
Grigg, R.; Sridharan, V.; York, M. Tetrahedron Lett. 1998, 39, 4139–4142. doi:10.1016/S0040-4039(98)00709-6
Return to citation in text: [1] -
de Vries, A. H. M.; Mulders, J. M. C. A.; Mommers, J. H. M.; Henderickx, H. J. W.; de Vries, J. G. Org. Lett. 2003, 5, 3285–3288. doi:10.1021/ol035184b
Return to citation in text: [1] -
McAlonan, H.; Montgomery, D.; Stevenson, P. J. Tetrahedron Lett. 1996, 37, 7151–7154. doi:10.1016/0040-4039(96)01564-X
Return to citation in text: [1] -
Bielawski, C. W.; Louie, J.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 12872–12873. doi:10.1021/ja001698j
Return to citation in text: [1] -
Leclerc, J.-P.; André, M.; Fagnou, K. J. Org. Chem. 2006, 71, 1711–1714. doi:10.1021/jo0523619
Return to citation in text: [1] -
Ibarguren, O.; Zakri, C.; Fouquet, E.; Felpin, F.-X. Tetrahedron Lett. 2009, 50, 5071–5074. doi:10.1016/j.tetlet.2009.06.084
Return to citation in text: [1] -
Fenster, M. D. B.; Dake, G. R. Org. Lett. 2003, 5, 4313–4316. doi:10.1021/ol035566h
Return to citation in text: [1] -
Yang, D.; Wong, M.-K.; Yip, Y.-C. J. Org. Chem. 1995, 60, 3887–3889. doi:10.1021/jo00117a046
Return to citation in text: [1] -
Kamenecka, T. M.; Danishefsky, S. J. Chem.–Eur. J. 2001, 7, 41–63. doi:10.1002/1521-3765(20010105)7:1<41::AID-CHEM41>3.0.CO;2-D
Return to citation in text: [1] [2] -
Li, S.; Yamamura, S. Tetrahedron 1998, 54, 8691–8710. doi:10.1016/S0040-4020(98)00479-7
Return to citation in text: [1] [2] -
Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Berti, G.; Marsili, A. Tetrahedron 1966, 22, 2977–2988. doi:10.1016/S0040-4020(01)82275-4
(see for example).
Return to citation in text: [1] -
Speckamp, W. N.; Moolenaar, M. J. Tetrahedron 2000, 56, 3817–3856. doi:10.1016/S0040-4020(00)00159-9
Return to citation in text: [1] -
Ungureanu, I.; Bologa, C.; Chayer, S.; Mann, A. Tetrahedron Lett. 1999, 40, 5315–5318. doi:10.1016/S0040-4039(99)01002-3
Return to citation in text: [1] -
Qiu, J.; Silverman, R. B. J. Med. Chem. 2000, 43, 706–720. doi:10.1021/jm9904755
Return to citation in text: [1] -
Blanchet, J.; Macklin, T.; Ang, P.; Metallinos, C.; Snieckus, V. J. Org. Chem. 2007, 72, 3199–3206. doi:10.1021/jo062385v
Return to citation in text: [1] -
Seto, H.; Fujioka, S.; Koshino, H.; Suenaga, T.; Yoshida, S.; Watanabe, T.; Takatsuto, S. J. Chem. Soc., Perkin Trans. 1 1998, 3355–3358. doi:10.1039/a805945d
Return to citation in text: [1]
| 20. | Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587 |
| 20. | Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587 |
| 20. | Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587 |
| 1. | Evans, P.; McCabe, T.; Morgan, B. S.; Reau, S. Org. Lett. 2005, 7, 43–46. doi:10.1021/ol0480123 |
| 2. | Evans, P. J. Org. Chem. 2007, 72, 1830–1833. doi:10.1021/jo062189o |
| 3. | Kelleher, S.; Muldoon, J.; Müller-Bunz, H.; Evans, P. Tetrahedron Lett. 2007, 48, 4733–4736. doi:10.1016/j.tetlet.2007.05.015 |
| 4. | Zeng, W.; Chemler, S. R. J. Org. Chem. 2008, 73, 6045–6047. doi:10.1021/jo801024h |
| 1. | Evans, P.; McCabe, T.; Morgan, B. S.; Reau, S. Org. Lett. 2005, 7, 43–46. doi:10.1021/ol0480123 |
| 2. | Evans, P. J. Org. Chem. 2007, 72, 1830–1833. doi:10.1021/jo062189o |
| 3. | Kelleher, S.; Muldoon, J.; Müller-Bunz, H.; Evans, P. Tetrahedron Lett. 2007, 48, 4733–4736. doi:10.1016/j.tetlet.2007.05.015 |
| 25. | Blanchet, J.; Macklin, T.; Ang, P.; Metallinos, C.; Snieckus, V. J. Org. Chem. 2007, 72, 3199–3206. doi:10.1021/jo062385v |
| 11. | de Vries, A. H. M.; Mulders, J. M. C. A.; Mommers, J. H. M.; Henderickx, H. J. W.; de Vries, J. G. Org. Lett. 2003, 5, 3285–3288. doi:10.1021/ol035184b |
| 26. | Seto, H.; Fujioka, S.; Koshino, H.; Suenaga, T.; Yoshida, S.; Watanabe, T.; Takatsuto, S. J. Chem. Soc., Perkin Trans. 1 1998, 3355–3358. doi:10.1039/a805945d |
| 1. | Evans, P.; McCabe, T.; Morgan, B. S.; Reau, S. Org. Lett. 2005, 7, 43–46. doi:10.1021/ol0480123 |
| 2. | Evans, P. J. Org. Chem. 2007, 72, 1830–1833. doi:10.1021/jo062189o |
| 3. | Kelleher, S.; Muldoon, J.; Müller-Bunz, H.; Evans, P. Tetrahedron Lett. 2007, 48, 4733–4736. doi:10.1016/j.tetlet.2007.05.015 |
| 10. | Grigg, R.; Sridharan, V.; York, M. Tetrahedron Lett. 1998, 39, 4139–4142. doi:10.1016/S0040-4039(98)00709-6 |
| 24. | Qiu, J.; Silverman, R. B. J. Med. Chem. 2000, 43, 706–720. doi:10.1021/jm9904755 |
| 5. |
Knowles, J. P.; Whiting, A. Org. Biomol. Chem. 2007, 5, 31–44. doi:10.1039/b611547k
(see for recent reviews). |
| 6. |
Zeni, G.; Larock, R. C. Chem. Rev. 2006, 106, 4644–4680. doi:10.1021/cr0683966
(see for recent reviews). |
| 7. |
Gibson, S. E.; Middleton, R. J. Contemp. Org. Synth. 1996, 3, 447–471. doi:10.1039/CO9960300447
(see for recent reviews). |
| 8. |
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
(see for recent reviews). |
| 9. |
Link, J. T. Org. React. 2002, 60, 157–534. doi:10.1002/0471264180.or060.02
(see for recent reviews). |
| 20. | Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587 |
| 18. | Kamenecka, T. M.; Danishefsky, S. J. Chem.–Eur. J. 2001, 7, 41–63. doi:10.1002/1521-3765(20010105)7:1<41::AID-CHEM41>3.0.CO;2-D |
| 19. | Li, S.; Yamamura, S. Tetrahedron 1998, 54, 8691–8710. doi:10.1016/S0040-4020(98)00479-7 |
| 18. | Kamenecka, T. M.; Danishefsky, S. J. Chem.–Eur. J. 2001, 7, 41–63. doi:10.1002/1521-3765(20010105)7:1<41::AID-CHEM41>3.0.CO;2-D |
| 19. | Li, S.; Yamamura, S. Tetrahedron 1998, 54, 8691–8710. doi:10.1016/S0040-4020(98)00479-7 |
| 21. |
Berti, G.; Marsili, A. Tetrahedron 1966, 22, 2977–2988. doi:10.1016/S0040-4020(01)82275-4
(see for example). |
| 16. | Fenster, M. D. B.; Dake, G. R. Org. Lett. 2003, 5, 4313–4316. doi:10.1021/ol035566h |
| 17. | Yang, D.; Wong, M.-K.; Yip, Y.-C. J. Org. Chem. 1995, 60, 3887–3889. doi:10.1021/jo00117a046 |
| 22. | Speckamp, W. N.; Moolenaar, M. J. Tetrahedron 2000, 56, 3817–3856. doi:10.1016/S0040-4020(00)00159-9 |
| 23. | Ungureanu, I.; Bologa, C.; Chayer, S.; Mann, A. Tetrahedron Lett. 1999, 40, 5315–5318. doi:10.1016/S0040-4039(99)01002-3 |
| 13. | Bielawski, C. W.; Louie, J.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 12872–12873. doi:10.1021/ja001698j |
| 14. | Leclerc, J.-P.; André, M.; Fagnou, K. J. Org. Chem. 2006, 71, 1711–1714. doi:10.1021/jo0523619 |
| 15. | Ibarguren, O.; Zakri, C.; Fouquet, E.; Felpin, F.-X. Tetrahedron Lett. 2009, 50, 5071–5074. doi:10.1016/j.tetlet.2009.06.084 |
| 20. | Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587 |
| 12. | McAlonan, H.; Montgomery, D.; Stevenson, P. J. Tetrahedron Lett. 1996, 37, 7151–7154. doi:10.1016/0040-4039(96)01564-X |
| 20. | Dura, R. D.; Paquette, L. A. J. Org. Chem. 2006, 71, 2456–2459. doi:10.1021/jo0526587 |
© 2009 Kelleher et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)