Abstract
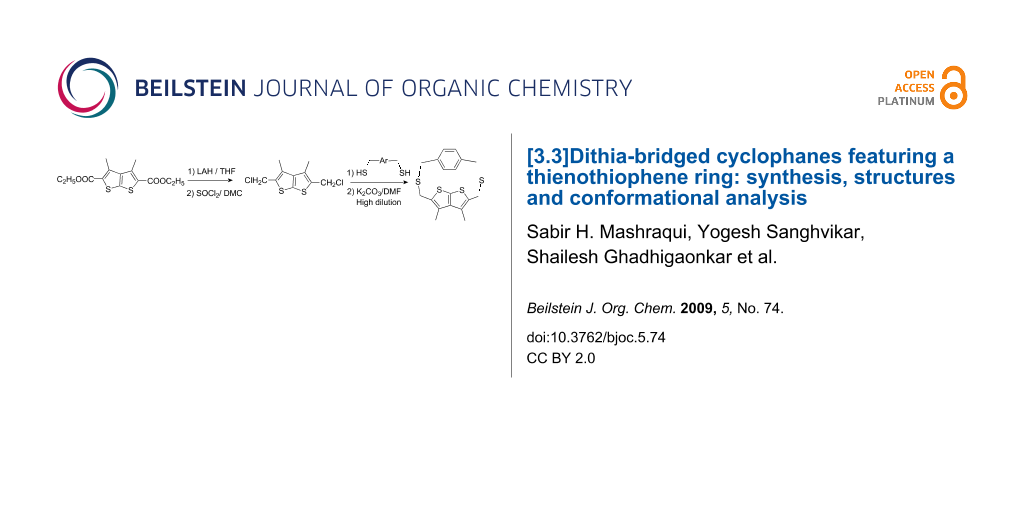
The synthesis of [3.3]dithia-bridged cyclophanes 7, 9 and 11 incorporating a fused heterocycle, thieno[2,3-b]thiophene is described. The structures are established by 1H NMR analysis and, in the case of 11, also by single crystal X-ray crystallography. Conformational analysis by variable temperature NMR suggests that cyclophanes 7, 9 and 11 exhibit conformationally rigid bridges and rings at least up to 130 °C. Energy minimization of 11 revealed anti-11 to be the most stable conformation. Although, the computed energy difference between the most stable conformation anti-11 and the next higher energy conformation syn-anti-11 is only 2.98 kJ/mol, it is intriguing that 11 does not exhibit thia-bridge inversion even at elevated temperatures.

Graphical Abstract
Introduction
The synthesis, molecular structures and conformational dynamics of short-bridged cyclophanes continue to engage interest in supramolecular chemistry [1-4]. The study of the conformational dynamics of cyclophanes has greatly enriched our fundamental understanding with regard to the structural features governing the energy barriers to ring inversion processes in these molecules [5,6]. Structural factors that have bearing on the conformational energy barriers of the ring and bridges in cyclophanes include the type the rings, nature of inner substituents and the length of the bridges encompassing the ring(s). Temperature dependent conformational behaviors of a diverse variety of [1.3]dithia-cyclophanes have been studied extensively and depending upon structural features both conformationally restricted and freely interconverting thia-bridged systems have been identified [7-17].
Cyclophanes incorporating simple heteronuclei such as furan, thiophene, pyridine, pyridazine and benzothiazole are abundantly known [1-3]. Despite the known structural diversity and complexity of cyclophanes, it is surprising that cyclophanes featuring fused heteronuclei have scarcely been studied [18-20]. Lately, we have been exploiting the thieno[2,3-b]thiophene ring, a fused heterocycle towards creating potentially interesting intramolecular charge transfer [21], nonlinear optics [22] and axially chiral systems [23]. We have also recently incorporated a thienothiophene ring within the cyclophane framework via its 3,4-positions, leading to the synthesis of meta type 1,3-dithia-bridged thienothiophenophanes 1–2 [24]. Cyclophanes 1–2 (Figure 1) have been found to exhibit thia-bridge inversion down to −50 °C with an estimated energy barrier >25 kJ mol−1. In the present work, we deemed it of interest to exploit the other available positions of the thienothiophene ring, namely the 2, 5 positions to access a new set of bridged [3.3]dithia-thienothiophenophanes 7, 9 and 11. Our main objective in the synthesis of these cyclophanes was to evaluate their structures and conformational properties.
![[1860-5397-5-74-1]](/bjoc/content/figures/1860-5397-5-74-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Conformationally flexible 3, 4-bridged dithia-thienothiophenophanes.
Figure 1: Conformationally flexible 3, 4-bridged dithia-thienothiophenophanes.
Results and Discussion
Synthesis of dithia-thienothiophenophanes 7, 9 and 11
2,5-Bis(chloromethyl)-3,4-dimethylthieno[2,3-b]thiophene (5) required as a precursor to construct cyclophanes 7, 9 and 11 was prepared as outlined in Scheme 1. The starting material, thienothiophene diester 3 was readily prepared in multigram quantities by a modification of our procedure [25]. Reduction of 3 with LiAlH4 in dry THF, followed by basic aqueous work-up gave diol 4 in 91% yield. The reaction of 4 with SOCl2 in dry dichloromethane (5–10 °C, 5 h), followed by solvent removal and crystallization from petroleum ether afforded dichloride 5 in 88% yield, m.p. 161–163 °C. The coupling reaction of 5 with m-xylenedithiol 6 was carried out in dry DMF/K2CO3 under high dilution conditions and under N2 atmosphere. The crude solid obtained on work-up was purified by column chromatography on SiO2 affording the desired meta dithia[3.3]cyclophane 7, m.p. 247–250 °C as a colorless solid in 57% yield. Likewise, the para dithia-bridged thienothiophenophanes 9 and 11 were prepared by coupling dichloride 5 with dithiols 8 and 10 respectively under the conditions described for 7. The crude products from these reactions were purified by column chromatography on SiO2 to furnish the corresponding dithia-cyclophanes 9 and 11 in 50% and 10% yields respectively.
![[1860-5397-5-74-i1]](/bjoc/content/inline/1860-5397-5-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic scheme for compounds 7, 9 and 11.
Scheme 1: Synthetic scheme for compounds 7, 9 and 11.
NMR assignments and conformational analysis of thienothiophenophanes 7, 9 and 11
The 1H NMR (300 MHz) spectrum of dithia-cyclophane 7 revealed two AB spin systems (4H each) in the range of δ 3.21–4.30 due to the geminal couplings of the bridged –CH2- protons. A singlet centered at δ 2.31 accounted for the C3/C4 methyl groups. The adjacent aromatic H4, H5 and H6 protons appeared as a multiplet (3H) centered at δ 7.32, whereas the isolated internal H2 proton appeared as a sharp singlet (1H) at δ 6.03. The higher field position of the internal H2 compared to the chemical shifts of H4-H5-H6 protons is presumably due to the shielding anisotropic effect of the facing thienothiophene ring.
The 1H NMR (700 MHz) spectrum of dithia-cyclophane 9 showed two sets of AB type spin systems, comprising doublets at δ 3.65 (2H, J = 15 Hz), 3.72 (2H, J = 14 Hz), 3.73 (2H, J = 15 Hz) and 4.08 (2H, J = 14 Hz). The appearance of two AB type spin systems implies that the methylene protons attached to one side of the bridge are magnetically identical with their respective counterparts on the other side of the bridge. A singlet located at δ 2.19 is due to the methyl protons, whereas the phenyl ring protons appear as sharp singlets at δ 6.84 and 6.95 (2H each). Appearance of two singlets for the para bridged phenyl ring implies that the two sides of the phenyl ring are subject to differing magnetic fields on account of the unsymmetrical nature of the facing thienothiophene ring.
The 1H NMR of 11 showed a 16 line resonance (δ 3.36 to 4.30) expected for the AB type coupling of its four unique sets of bridge methylene protons. Each of the −CH3 groups and −OCH3 groups appear as separate singlets observed at δ 2.20, 2.31 and 3.75, 3.82, respectively. The nonequivalence of otherwise chemically identical pair of −CH3 as well as −OCH3 group of protons can be attributed to the differing magnetic environments around these groups created by the facing thienothiophene and the phenyl rings. Although, in analogy to the −CH3 and −OCH3 protons, we expected the phenyl ring protons to also appear as two separate singlets, however in practice we observe only one singlet at δ 6.80 (2H). This may presumably be due to their coincidental chemical shift equivalence.
The bridge −CH2− protons in dithia-bridged cyclophanes 7, 9 and 11 resonate as geminally coupled AB systems. From these results, we infer that the bridge inversions in these molecules are restricted on the NMR time scale at ambient temperatures. Furthermore, since phenyl ring protons in 9 and methoxy protons in 11 resonate at two different chemical shifts, we can conclude that aryl ring rotations in these molecules also seem to be conformationally restricted. Higher temperatures might surpass the conformational energy barrier, allowing the molecules to undergo free bridge and ring inversions [26]. In the event of free bridge rotations, the geminally coupled systems would coalesce into two sharp singlets corresponding to the two different sets of the bridged −CH2− protons. On the other hand, ring rotation in 9 and 11 would convert separate signals for the phenyl ring and methoxyl protons into singlets due to the fast interconversion of ring conformers. Accordingly, we recorded variable temperature NMR spectra of dithia-cyclophanes 7, 9 and 11 at different intervals from room temperature to 130 °C in DMSO-d6. However, no changes were noticeable in the multiplicities of the bridge −CH2− protons with the aryl ring and methoxy protons also retaining their separate chemical shift positions. These observations suggest the absence of bridge flippings and ring rotations in these systems at least up to 130 °C with an estimated conformational energy barrier of >100 kJ mol−1 [27]. The presence of substituents on the thienothiophene ring (as well as on the phenyl ring in 11) and the absence of conformational inversion endow these with molecular chirality rendering them potentially resolvable.
Temperature dependent conformational behaviors have been reported for a number of unsubstituted [1.3] dithia-cyclophanes [5,6]. However, replacing internal hydrogen atoms of the thienothiophene with a bulky substituent viz. −CH3 in [1.3]dithia- and related cyclophanes raises the conformational energy barriers sufficiently to render such systems conformationally immobile [28-31]. By analogy, it is reasonable to assume that the conformational restrictions of the bridges and rings observed in the presently synthesized cyclophanes could at least in part be due to the presence of methyl substituents on the thienothiophene rings in 7 and 9 and both methyl and methoxyl substituents for case of 11.
Single X-ray Crystal structure and conformational search of 11
The single-crystal structure of 11 was deduced by X-ray crystallography [32]. The space group of the molecular structure is P21/n, meaning the point group is 2/m. An ORTEP drawing is shown in Figure 2 and the relevant crystallographic parameters are compiled in Table 1. The torsion angle C18–C14–C15–C16 and C10–C11–C12–C13 are −174.40(13)° and −175.80(13)° indicating that the benzene ring is slightly inwardly bend. The dihedral angles C8–C6–C4–C5 and C5–C4–C2–C1 are −2.00(19)° and 2.85(19)°, respectively; thereby implying that thienothiophene ring is also slightly inwardly bent. The thia bridges are anti oriented with respect to one another, and so are the two methoxyl groups. The angle of 104.13° for C10–S3–C9 is slightly larger than 101.13° for C18–S4–C17, presumably to minimized steric interaction between the methoxyl and methyl substituents on either sides of the bridges. The transannular distances between the bridgehead carbons C11–C6 and C14–C2 are 3.37 Å and 3.32 Å, respectively and the phenyl and thienothiophene rings are stacked with the former slightly displaced from the center of the latter.
![[1860-5397-5-74-2]](/bjoc/content/figures/1860-5397-5-74-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP plot of the crystal structure of 11. Important parameters: Bond length (Å) : C8–C9 = 1.492, C1–C7 = 1.497, C10–C11 = 1.517, C14–C18 = 1.522; Bond Angles (deg): C4, C6, C7 = 124.70, C1, C2, C3 = 125.03, C10, C11, C12 = 120.31, C13, C14, C18 = 123.14, C10, S3, C9 = 104.13; C18, S4, C17 = 101.14, C16, C11, C10 = 121.12 C15, C14, C18 = 118.43; Dihedral Angles (deg): C18, C14, C15, C16 = −174.40(13), C10, C11, C12, C13 = −175.43(14), C10, C11, C16, C15 = 178.80(13), C15, C16, C11, C12 = −2.7(2), C12, C13, C14, C15 = −3.1(2), C8, C6, C4, C6 = 168.50(16), C1, C2, C4, C6 = −167.65(16), C5, C4, C2, C1 = 5.29.
Figure 2: ORTEP plot of the crystal structure of 11. Important parameters: Bond length (Å) : C8–C9 = 1.492, C...
Table 1: Summary of crystallographic data and refinement details.
| Empirical formula | C20H22O2S4 |
|---|---|
| Formula mass | 422.66 |
| Crystal color and habit | colorless, block |
| Crystal size [nm] | 0.23 × 0.21 × 0.19 |
| Crystal system | monoclinic |
| a [Å] | 8.2943(5) |
| b [Å] | 13.8204(9) |
| c [Å] | 16.679(1) |
| β [°] | 97.690(1) |
| V [Å3] | 1894.7(2) |
| ρcalcd. [g/cm3] | 1.482 |
| F(000) | 888 |
| μ [cm−1] | 5.14 (Mo-Kα) |
| 2θmax [°] | 55.0 (I > 3.0σ) |
| No. of reflections | 4039 |
In order to evaluate the energy minimized conformations for 11, we performed the conformational analysis by random search Monte Carlo method [33] and optimized by PRCG Conjugated Gradient molecular mechanics minimization using the Macromodel version 5.5 program [34], MM2 force field [35] and GB/SA chloroform solvation [36]. Two thousand conformations were generated from which we have selected three of the most stable conformations within 12.55 kJ/mol from the global minimum.
![[1860-5397-5-74-i2]](/bjoc/content/inline/1860-5397-5-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Computed three most stable conformations of 11 and their calculated energies.
Scheme 2: Computed three most stable conformations of 11 and their calculated energies.
As depicted in Scheme 2, the notable difference between these conformations is the relative orientation of the bridging sulfur atoms with respect to the thienothiophene ring. The conformation designated as anti-11 is unsymmetrical with the bridging sulfur atoms being anti-oriented i.e. facing away from each other. The conformations syn-anti-11 and syn-syn-11 both have their bridge ‘S’ atoms symmetric about a plane bisecting the two rings, though they are not symmetric to the plane. Both conformations, syn-anti-11 and syn-syn-11 possess syn-oriented sulfur bridges, however: for the former the bridging sulfur atoms are pointing away from the sulfur atoms of the thienothiophene ring, while for the latter, the bridging sulfur atoms and the thienothiophene ring sulfur atoms are facing the same side. The key dihedral angles of these conformations together with those of 11 derived from the X-ray data are compiled in Table 2.
Table 2: Selected computed dihedral angles (deg) of anti-11, syn-anti-11, and syn-syn-11 and those of 11 derived from the X-ray crystallography.
| anti-11 | syn-anti-11 | syn-syn-11 | 11 | |
|---|---|---|---|---|
| E in kJ/mol | −96.50 | −93.52 | −83.95 | – |
| ΔE in kJ/mol | 0.00 | 2.98 | 12.55 | – |
| S2–C5–S1–C1 | −162.7° | +161.0° | −162.9° | 168.61° (13) |
| C5–S1–C1–C17 | +169.8° | −168.5° | +170.3° | −165.12° (12) |
| S1–C1–C17–S4 | −61.3° | +51.8° | −120.4° | 73.25° (13) |
| C1–C17–S4–C18 | −67.0° | +71.9° | +63.0° | 56.29° (12) |
| C17–S4–C18–C14 | +78.9° | −69.8° | −74.9° | −85.25° (12) |
| C11–C10–S3–C9 | +82.9° | +80.2° | +82.5° | −78.54° (13) |
| C10–S3–C9–C8 | −58.2° | −68.3° | −60.3° | 49.78° (13) |
| S3–C9–C8–S2 | +113.3° | −61.8° | +112.5° | −106.16° (12) |
| C9–C8–S2–C5 | −169.8° | +169.2° | −168.5° | 169.94° (13) |
| C8–S2–C5–S1 | +163.6 | −161.7 | +162.4 | −169.57 (13) |
The conformation anti-11 is the lowest energy structure with ca. E of −96.50 kJ/mol, while syn-anti-11 and syn-syn-11 are of relatively higher energy by ca. 2.98 and 12.55 kJ/mol, respectively. Inspection of Table 2 reveals that while some of the computed dihedral angles of the three most stable conformations are quite close to each other, the computed dihedral angles viz. S1–C1–C17–S4, C17–S4–C18–C14, C10–S3–C9–C8 and C8–S2–C5–S1 for the most stable anti-11 are broadly similar to those of the cyclophane 11 derived from the X-ray data. Consequently, there is good correspondence between the X-ray crystal structure and the most stable conformation anti-11 computed from the conformation energy minimization. Although, the ΔE between the most stable conformation anti-11 and the next higher energy conformation syn-anti-11 is only 2.98 kJ/mol, it is intriguing that 11 does not exhibit thia-bridge inversion even at elevated temperatures. The flipping of the thia-bridges in 11 as well as other cyclophanes described in the preceding section could be inhibited if the transition state structures have free energies of activation >100 kJ/mol [5]. This aspect is currently being examined using molecular modeling approach.
Conclusion
In conclusion, we have reported the synthesis and structures of [3.3]dithia-bridged thienothiophenophanes 7, 9, and 11. Variable temperature 1H NMR analysis revealed the absence of bridge flipping up to 130°C in these molecules. Phenyl ring rotation in cyclophanes 9 and 11 is also ruled out since separate singlets associated with phenyl protons (in case of 11, also the –OCH3) failed to coalesce up to 130 °C. The structure of 11 was also deduced by a single crystal X-ray crystallography. The most stable conformation anti-11 derived from conformational minimization possessed broad structural similarity with that of the X-ray crystal structure.
Experimental
The chemicals and spectroscopic grade solvents were purchased from S.D.F. Chemicals (India) and Aldrich and used as received. IR spectra were recorded on a Shimadzu FTIR-420 spectrophotometer. 1H NMR spectra were scanned in CDCl3 using a Bruker 300 or 700 MHz spectrometer with TMS as an internal standard. Elemental analyses were done on a Carlo Erba instrument EA-1108 Elemental analyzer.
Preparation of 3,4-dimethyl-2,5-bis(hydroxymethyl)thieno[2,3-b]thiophene (4)
A solution of diester thienothiophene 3 (1.56 g, 5 mmol) dissolved in dry THF (20 ml) was added to a slurry of LiAlH4 (950 mg, 25 mmol) prepared in dry THF (50 ml) at room temperature over a period of 1 h. After stirring for 24 h at room temperature, the reaction mixture was cooled in an ice-salt bath and hydrolyzed by successive additions of water (1 ml), 15% sodium hydroxide (1 ml) and water (2.5 ml) with vigorous stirring. The reaction mixture was then filtered through celite and the filtered cake washed thoroughly with dry THF. The filtrate was concentrated to give diol 4 as a colorless solid (1.03 g, 91% yield); mp: 156–158 °C; IR (KBr) cm−1: 3250, 2921, 2861, 1525, 1443, 1418, 1399, 1353, 1234, 1191, 1125, 1113, 1003, 949, 700. 1H NMR (DMSO, 300MHz) δ : 5.20(s, 2H, –OH), 4.60(s, 4H, −CH2−), 2.40(s, 6H, −CH3). Anal. Calcd for C10H12O2S2: C, 52.63; H, 5.26; S, 28.07. Found C, 52.78; H, 5.49; S, 28.23.
Preparation of 3,4-dimethyl-2,5-bis(chloromethyl)thieno[2,3-b]thiophene (5)
Diol 4 (1.14 g, 5 mmol) was stirred in dry dichloromethane and freshly distilled thionyl chloride (1.0 ml, 13.6 mmol) was added dropwise at 5–10 °C. The reaction mixture was allowed to warm to room temperature over 5 h. The reaction mixture was then concentrated and the obtained brown solid was boiled with light petroleum ether. The supernatant was transferred to a dry flask and the clear solution refrigerated overnight to furnish dichloride 5 as white–brownish solid (1.15 g, 88% yield); mp 161–163 °C; IR (KBr) cm−1: 2923, 1520, 1436, 1422, 1395, 1261, 1174, 1131, 1112, 862, 811, 673. 1H NMR (CDCl3, 300 MHz) δ: 4.70 (s, 4 H, –CH2), 2.50 (s, 6H, –CH3). Anal. Calcd for C10H10Cl2S2: C, 45.28; H, 37.74, S, 24.15, Cl, 26.42 Found C, 45.40; H, 37.57; S, 24.28, Cl, 26.35.
Preparation of dithia[3]metacyclo[3](2,5)thienothiophenophane 7
A solution of dichloride 5 (265 mg, 1 mmol) and 1,3-bis(mercaptomethyl)benzene (6) (170 mg, 1 mmol) in dry DMF (50 ml) was added dropwise to a stirred mixture of dry DMF (100 ml) containing anhyd. K2CO3 (250 mg) at 60–70 °C over 8 h under N2 atmosphere. The reaction mixture was further stirred at this temperature for 16 h. It was then allowed to cool and filtered through a pad of celite. The filtrate was concentrated in vacuum to give a dark–brown colored residue. The residual solid was extracted in chloroform, washed with water and dried over anhyd. Na2SO4. The crude product obtained on concentration was purified by column chromatography on SiO2 using petroleum ether–chloroform (2:1) as an eluant. The cyclophane 7 was obtained as a colorless solid, (207 mg, 57%), m.p. 247–250 °C; IR (KBr) cm−1: 3008, 2947, 2907, 1583, 1482, 1428, 1396, 1228, 1215, 1082, 1009, 978, 780, 711. 1H NMR (CDCl3, 300 MHz) δ : 7.02 (s, 3H, Ph–H), 6.03 (s, 1H, Ph–H), 4.30 (d, 2H, J = 16Hz, –CH2-thienothiophene), 3.82 (d, 2H, J = 16Hz, −CH2-thienothiophene), 3.56 (d, 2H, J = 16 Hz, −CH2-phenyl), 3.21 (d, 2H, J = 16Hz, −CH2-phenyl), 2.31(s, 6H, −CH3). Mass : m/e 362. Anal. Calcd for C18H18S4: C, 59.67; H, 4.97; S, 35.36. Found C, 59.51; H, 4.93; S, 35.61.
Preparation of dithia[3]paracyclo[3](2,5)thienothiophenophane 9
Dichloride 5 (265 mg, 1 mmol) and 1,4-bis-(mercaptomethyl)benzene (8) (170 mg, 1 mmol) were coupled as described for 7. The crude product was chromatographed on SiO2 column using petroleum ether–chloroform (2:1) as an eluant to afford cyclophane 9 as a colorless solid (180 mg, 50%); mp 210–212 °C; IR (KBr) cm−1 : 2922, 1525, 1504, 1445, 1414, 1397, 1376, 1215, 1122, 1101, 1006, 892, 838, 791, 776, 722. 1H NMR (700 MHz, CDCl3) δ: 6.95 (s, 2H, Ph–H), 6.84 (s, 2H, Ph–H), 4.08 (2H, J = 14 Hz), 3.73 (2H, J = 15 Hz), 3.72 (2H, J = 14 Hz), 3.65 (2H, J = 15 Hz), 2.19 (s, 6H, −CH3). 13C NMR (300 MHz, CDCl3) δ: 147.37, 136.20, 135.11, 133.80, 129.08, 126.07, 125.22, 36.07, 30.87, 13.72. Mass: m/e 362. Anal. Calcd for C18H18S4: C, 59.67; H, 4.97; S, 35.36. Found C, 59.73; H, 4.81; S, 35.49.
Preparation of dimethoxydithia[3]paracyclo[3](2,5)thienothiophenophane 11
Dichloride 5 (265 mg, 1 mmol) and 2,5-dimethoxy-1,4-bis(mercaptomethyl)benzene (10) (156 mg, 1 mmol) were coupled as described for 7. The crude product was chromatographed on SiO2 using petroleum ether–chloroform (2:1) as an eluant to afford cyclophane 11 as a colorless solid (23 mg, 10% yield); mp 252–253 °C. IR (KBr) cm−1: 2988, 2950, 2836, 1606, 1576, 1492, 1459, 1446, 1409, 1308, 1231, 1196, 1042, 1007, 890, 864, 769, 749. 1H NMR (300 MHz, CDCl3) δ : 6.80 (s, 2H, Ph–H), 4.30 (d, 1H, J = 14 Hz, –CH2), 4.14 (d, 1H, J = 17 Hz, –CH2), 4.02 (d, 1H, J = 18 Hz, –CH2), 3.9 (d, 1H, J = 17 Hz, –CH2), 3.81 (d, 1H, J = 14 Hz, –CH2), 3.58 (d, 1H, J = 15 Hz, –CH2), 3.44 (d, 1H, J = 18 Hz, –CH2), 3.36 (d, 1H, J = 14 Hz, –CH2), 3.75 (s, 3H, –OCH3), 3.80 (s, 3H, –OCH3), 2.22 (s, 3H, –CH3), 2.29 (s, 3H, –CH3). Mass: m/e 422. Anal. Calcd for C20H22O2S4: C, 56.87; H, 5.21; S, 30.33. Found C, 56.94; H, 5.45; S, 30.19.
References
-
Keehn, P. M.; Rosenfeld, S. M., Eds. Cyclophanes; Academic Press: New York, 1983; Vol. I and II.
Return to citation in text: [1] [2] -
Vogtle, F. Cyclophane Chemistry; Wiley-VCH: Chichester, 1993.
Return to citation in text: [1] [2] -
Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, 2004.
Return to citation in text: [1] [2] -
Lehn, J.-M. Supramolecular Chemistry. Concepts and Perspectives; Wiley-VCH: Weinheim, 1995.
Return to citation in text: [1] -
Mitchell, R. H. In Cyclophanes; Keehn, P.; M and Rosenfeld, S. M., Eds.; Academic Press: New York, 1983; Vol. 1, pp 239–310.
Return to citation in text: [1] [2] [3] -
Ernst, L. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 37, 47–190. doi:10.1016/S0079-6565(00)00022-4
Return to citation in text: [1] [2] -
Mitchell, R. H.; Weerawarna, K. S.; Bushnell, G. W. Tetrahedron Lett. 1984, 25, 907–910. doi:10.1016/S0040-4039(01)80059-9
Return to citation in text: [1] -
Xu, J.-W.; Lin, T.-T.; Lai, Y.-H. Tetrahedron 2005, 61, 2431–2440. doi:10.1016/j.tet.2005.01.027
Return to citation in text: [1] -
Bodwell, G. J.; Ernst, L.; Hopf, H.; Jones, P. G.; McNally, J. P.; Schomburg, D. Chem. Ber. 1990, 123, 2381–2386. doi:10.1002/cber.19901231221
Return to citation in text: [1] -
Haenel, M. W. Chem. Ber. 1982, 115, 1425–1436. doi:10.1002/cber.19821150421
Return to citation in text: [1] -
Yamato, T.; Miyazawa, A.; Tashiro, M. J. Chem. Soc., Perkin Trans. 1 1993, 3127–3137. doi:10.1039/P19930003127
Return to citation in text: [1] -
Ernst, L.; Ibrom, K. Angew. Chem. 1995, 34, 1881–1882. doi:10.1002/anie.199518811
Return to citation in text: [1] -
Fukazawa, Y.; Ohta, E.; Nakabai, T.; Usui, S. Chem. Lett. 1987, 12, 2345–2346.
Return to citation in text: [1] -
Lai, Y.-H.; Ang, S.-G.; Wong, S.-Y. Tetrahedron Lett. 1997, 38, 2553–2556. doi:10.1016/S0040-4039(97)00399-7
Return to citation in text: [1] -
Mashraqui, S. H.; Keehn, P. M. J. Org. Chem. 1983, 48, 1341–1344. doi:10.1021/jo00156a037
Return to citation in text: [1] -
Mashraqui, S. H.; Sanghvikar, Y. S.; Meetsma, A. Tetrahedron Lett. 2006, 47, 5599–5602. doi:10.1016/j.tetlet.2006.05.098
Return to citation in text: [1] -
Mashraqui, S. H.; Kumar, S.; Dau, E. T. H. J. Mol. Struct. 2004, 697, 221–230. doi:10.1016/j.molstruc.2004.04.014
Return to citation in text: [1] -
Ueda, T.; Kanomata, N.; Machida, H. Org. Lett. 2005, 7, 2365–2368. doi:10.1021/ol0506258
Return to citation in text: [1] -
Butler, D. N.; Shang, M.; Warrener, R. N. Tetrahedron Lett. 2000, 41, 5985–5989. doi:10.1016/S0040-4039(00)00956-4
Return to citation in text: [1] -
Grimme, S. Chem.–Eur. J. 2004, 10, 3423–3429. doi:10.1002/chem.200400091
Return to citation in text: [1] -
Mashraqui, S. H.; Ashraf, M.; Hariharasubrahmanian, H.; Kellogg, R. M.; Meetsma, A. J. Mol. Struct. 2004, 689, 107–113. doi:10.1016/j.molstruc.2003.10.026
Return to citation in text: [1] -
Mashraqui, S. H.; Ghadigaonkar, S.; Ashraf, M.; SriRanjini, A.; Ghosh, S.; Das, P. K. Tetrahedron 2007, 63, 10011–10017. doi:10.1016/j.tet.2007.07.059
Return to citation in text: [1] -
Mashraqui, S. H.; Sanghvikar, Y.; Ashraf, M.; Kumar, S.; Trân Huu Dâub, E. Tetrahedron 2005, 61, 3507–3513. doi:10.1016/j.tet.2005.01.123
Return to citation in text: [1] -
Mashraqui, S. H.; Sanghvikar, Y. S.; Meetsma, A. Tetrahedron Lett. 2006, 47, 5599–5602. doi:10.1016/j.tetlet.2006.05.098
Return to citation in text: [1] -
Mashraqui, S. H.; Hariharasubramanian, H.; Kumar, S. Synthesis 1999, 12, 2030–2032. doi:10.1055/s-1999-3631
Return to citation in text: [1] -
Oki, M. Applications of Dynamic NMR Spectroscopy to Organic Chemistry. In Methods in Stereochemical Analysis; Marchand, A. P., Ed.; VCH Publishers: Deerfield Beach, Florida, 1985; Vol. 4, pp 234 ff.
Return to citation in text: [1] -
Sato, T.; Wakabayashi, H. M.; Kainosho, K. M. Tetrahedron 1971, 27, 2737–2755. doi:10.1016/S0040-4020(01)98064-0
Return to citation in text: [1] -
Lai, Y.-H.; Yap, A. H.-T. J. Chem. Soc., Perkin Trans. 2 1993, 7, 1373–1377. doi:10.1039/p29930001373
Return to citation in text: [1] -
Lai, Y.-H.; Yap, A. H.-T.; Novak, I. J. Org. Chem. 1994, 59, 3381–3385. doi:10.1021/jo00091a027
Return to citation in text: [1] -
Sherrod, S. A.; Boekelheide, V. J. Am. Chem. Soc. 1972, 94, 5513–5515. doi:10.1021/ja00770a066
Return to citation in text: [1] -
Staab, H. A.; Herz, C. P. Angew. Chem., Int. Ed. Engl. 1977, 16, 799–801. doi:10.1002/anie.197707991
Return to citation in text: [1] -
The X-ray crystal structure data have been submitted to the Cambridge Database as a file CCDC-297142.
Return to citation in text: [1] -
Chang, G.; Guida, W. C.; Still, W. C. J. Am. Chem. Soc. 1989, 111, 4379–4386. doi:10.1021/ja00194a035
Return to citation in text: [1] -
Mohamadi, F.; Richards, N. J. G.; Guida, W. C.; Liskamp, R.; Lipton, M. C.; Caufield, M.; Chang, G.; Hendrickson, T.; Still, W. C. J. Comput. Chem. 1990, 11, 440–467. doi:10.1002/jcc.540110405
Return to citation in text: [1] -
Allinger, N. L. J. Am. Chem. Soc. 1977, 99, 8127–8134. doi:10.1021/ja00467a001
Return to citation in text: [1] -
Still, W. C.; Tempczyk, A.; Hawley, R. C.; Hendrickson, T. J. Am. Chem. Soc. 1990, 112, 6127–6129. doi:10.1021/ja00172a038
Return to citation in text: [1]
| 36. | Still, W. C.; Tempczyk, A.; Hawley, R. C.; Hendrickson, T. J. Am. Chem. Soc. 1990, 112, 6127–6129. doi:10.1021/ja00172a038 |
| 34. | Mohamadi, F.; Richards, N. J. G.; Guida, W. C.; Liskamp, R.; Lipton, M. C.; Caufield, M.; Chang, G.; Hendrickson, T.; Still, W. C. J. Comput. Chem. 1990, 11, 440–467. doi:10.1002/jcc.540110405 |
| 35. | Allinger, N. L. J. Am. Chem. Soc. 1977, 99, 8127–8134. doi:10.1021/ja00467a001 |
| 1. | Keehn, P. M.; Rosenfeld, S. M., Eds. Cyclophanes; Academic Press: New York, 1983; Vol. I and II. |
| 2. | Vogtle, F. Cyclophane Chemistry; Wiley-VCH: Chichester, 1993. |
| 3. | Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, 2004. |
| 4. | Lehn, J.-M. Supramolecular Chemistry. Concepts and Perspectives; Wiley-VCH: Weinheim, 1995. |
| 18. | Ueda, T.; Kanomata, N.; Machida, H. Org. Lett. 2005, 7, 2365–2368. doi:10.1021/ol0506258 |
| 19. | Butler, D. N.; Shang, M.; Warrener, R. N. Tetrahedron Lett. 2000, 41, 5985–5989. doi:10.1016/S0040-4039(00)00956-4 |
| 20. | Grimme, S. Chem.–Eur. J. 2004, 10, 3423–3429. doi:10.1002/chem.200400091 |
| 32. | The X-ray crystal structure data have been submitted to the Cambridge Database as a file CCDC-297142. |
| 1. | Keehn, P. M.; Rosenfeld, S. M., Eds. Cyclophanes; Academic Press: New York, 1983; Vol. I and II. |
| 2. | Vogtle, F. Cyclophane Chemistry; Wiley-VCH: Chichester, 1993. |
| 3. | Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, 2004. |
| 33. | Chang, G.; Guida, W. C.; Still, W. C. J. Am. Chem. Soc. 1989, 111, 4379–4386. doi:10.1021/ja00194a035 |
| 7. | Mitchell, R. H.; Weerawarna, K. S.; Bushnell, G. W. Tetrahedron Lett. 1984, 25, 907–910. doi:10.1016/S0040-4039(01)80059-9 |
| 8. | Xu, J.-W.; Lin, T.-T.; Lai, Y.-H. Tetrahedron 2005, 61, 2431–2440. doi:10.1016/j.tet.2005.01.027 |
| 9. | Bodwell, G. J.; Ernst, L.; Hopf, H.; Jones, P. G.; McNally, J. P.; Schomburg, D. Chem. Ber. 1990, 123, 2381–2386. doi:10.1002/cber.19901231221 |
| 10. | Haenel, M. W. Chem. Ber. 1982, 115, 1425–1436. doi:10.1002/cber.19821150421 |
| 11. | Yamato, T.; Miyazawa, A.; Tashiro, M. J. Chem. Soc., Perkin Trans. 1 1993, 3127–3137. doi:10.1039/P19930003127 |
| 12. | Ernst, L.; Ibrom, K. Angew. Chem. 1995, 34, 1881–1882. doi:10.1002/anie.199518811 |
| 13. | Fukazawa, Y.; Ohta, E.; Nakabai, T.; Usui, S. Chem. Lett. 1987, 12, 2345–2346. |
| 14. | Lai, Y.-H.; Ang, S.-G.; Wong, S.-Y. Tetrahedron Lett. 1997, 38, 2553–2556. doi:10.1016/S0040-4039(97)00399-7 |
| 15. | Mashraqui, S. H.; Keehn, P. M. J. Org. Chem. 1983, 48, 1341–1344. doi:10.1021/jo00156a037 |
| 16. | Mashraqui, S. H.; Sanghvikar, Y. S.; Meetsma, A. Tetrahedron Lett. 2006, 47, 5599–5602. doi:10.1016/j.tetlet.2006.05.098 |
| 17. | Mashraqui, S. H.; Kumar, S.; Dau, E. T. H. J. Mol. Struct. 2004, 697, 221–230. doi:10.1016/j.molstruc.2004.04.014 |
| 5. | Mitchell, R. H. In Cyclophanes; Keehn, P.; M and Rosenfeld, S. M., Eds.; Academic Press: New York, 1983; Vol. 1, pp 239–310. |
| 6. | Ernst, L. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 37, 47–190. doi:10.1016/S0079-6565(00)00022-4 |
| 5. | Mitchell, R. H. In Cyclophanes; Keehn, P.; M and Rosenfeld, S. M., Eds.; Academic Press: New York, 1983; Vol. 1, pp 239–310. |
| 6. | Ernst, L. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 37, 47–190. doi:10.1016/S0079-6565(00)00022-4 |
| 28. | Lai, Y.-H.; Yap, A. H.-T. J. Chem. Soc., Perkin Trans. 2 1993, 7, 1373–1377. doi:10.1039/p29930001373 |
| 29. | Lai, Y.-H.; Yap, A. H.-T.; Novak, I. J. Org. Chem. 1994, 59, 3381–3385. doi:10.1021/jo00091a027 |
| 30. | Sherrod, S. A.; Boekelheide, V. J. Am. Chem. Soc. 1972, 94, 5513–5515. doi:10.1021/ja00770a066 |
| 31. | Staab, H. A.; Herz, C. P. Angew. Chem., Int. Ed. Engl. 1977, 16, 799–801. doi:10.1002/anie.197707991 |
| 24. | Mashraqui, S. H.; Sanghvikar, Y. S.; Meetsma, A. Tetrahedron Lett. 2006, 47, 5599–5602. doi:10.1016/j.tetlet.2006.05.098 |
| 26. | Oki, M. Applications of Dynamic NMR Spectroscopy to Organic Chemistry. In Methods in Stereochemical Analysis; Marchand, A. P., Ed.; VCH Publishers: Deerfield Beach, Florida, 1985; Vol. 4, pp 234 ff. |
| 23. | Mashraqui, S. H.; Sanghvikar, Y.; Ashraf, M.; Kumar, S.; Trân Huu Dâub, E. Tetrahedron 2005, 61, 3507–3513. doi:10.1016/j.tet.2005.01.123 |
| 27. | Sato, T.; Wakabayashi, H. M.; Kainosho, K. M. Tetrahedron 1971, 27, 2737–2755. doi:10.1016/S0040-4020(01)98064-0 |
| 22. | Mashraqui, S. H.; Ghadigaonkar, S.; Ashraf, M.; SriRanjini, A.; Ghosh, S.; Das, P. K. Tetrahedron 2007, 63, 10011–10017. doi:10.1016/j.tet.2007.07.059 |
| 5. | Mitchell, R. H. In Cyclophanes; Keehn, P.; M and Rosenfeld, S. M., Eds.; Academic Press: New York, 1983; Vol. 1, pp 239–310. |
| 21. | Mashraqui, S. H.; Ashraf, M.; Hariharasubrahmanian, H.; Kellogg, R. M.; Meetsma, A. J. Mol. Struct. 2004, 689, 107–113. doi:10.1016/j.molstruc.2003.10.026 |
| 25. | Mashraqui, S. H.; Hariharasubramanian, H.; Kumar, S. Synthesis 1999, 12, 2030–2032. doi:10.1055/s-1999-3631 |
© 2009 Mashraqui et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)