Abstract
The effect of the phenyl–alkyl connecting group on mesogenic properties of several series of isostructural compounds containing p-carborane (A and B), bicyclo[2.2.2]octane (C), and benzene (D) was investigated using thermal and optical methods. Results demonstrated that mesophase stability in the series containing A–D follows the order (Alk)CH2CH2– < (Alk)OOC– < (Alk)CH2O– < (Alk)COO–. Surprisingly, the connecting groups (Alk)CH2CH2– and (Alk)OOC– destabilize the mesophase significantly stronger for carboranes (A and B) than for carbocyclic derivatives (C and D). Analysis indicates that this effect may have quadrupolar and conformational origin.

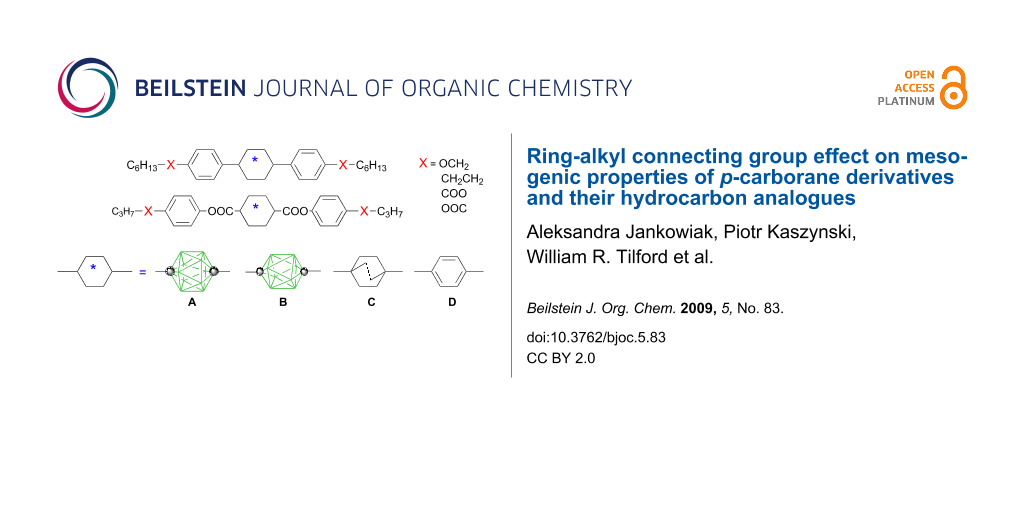
Graphical Abstract
Introduction
During the past decade, we have been investigating mesogenic derivatives of p-carboranes A and B (Figure 1) in the context of fundamental and applied studies of liquid crystals and development of new materials for electrooptical applications [1-23]. p-Carboranes belong to an extensive family of closo-boranes and are characterized by 3-dimensional σ-aromaticity and high-order symmetry axis [23]. Therefore, it is of interest to understand how the electronic properties of the two clusters and their unusual molecular symmetry and size affect bulk properties of mesogens. Through extensive comparison of isostructural mesogenic derivatives of p-carboranes (A and B), bicyclo[2.2.2]octane (C), and benzene (D), we have been probing fundamental aspects of structure-property relationships in liquid crystals such as the effect of conformational properties [1,2], the structure of the linking group [5], and tail fluorination [18,19] on mesophase stability, and also the effectiveness of shielding of a lateral substituent [8,16,20] and chirality transfer phenomena [17]. Results of these studies are important for the design of new materials and optimizing of their properties for applications.
![[1860-5397-5-83-1]](/bjoc/content/figures/1860-5397-5-83-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The molecular structures of 1,12-dicarba-closo-dodecaborane (12-vertex p-carborane, A) and 1,10-dicarba-closo-decaborane (10-vertex p-carborane, B). Each vertex corresponds to a BH fragment and the sphere represents a carbon atom.
Figure 1: The molecular structures of 1,12-dicarba-closo-dodecaborane (12-vertex p-carborane, A) and 1,10-dic...
During our investigation of structurally related series of mesogens containing rings A–D (Figure 2), it became apparent that the benzene ring–alkyl chain connection has a distinctly different impact on phase stability in derivatives of p-carborane (A) than in their isostructural carbocycles. For instance, a larger stabilization of the nematic phase, upon CH2→O replacement, was observed in p-carborane mesogens relative to benzene analogues. Thus, in series 1–4, the nematic phase is stabilized by about 14 K more for the pairs 1A/2A and 3A/4A, than for terphenyl (D) and bicyclo[2.2.2]octane (C) analogues (Figure 2). Similarly high values for phase stabilization of about 34 K are observed in pairs of alkyl and alkoxy dioxane derivatives 5[n] and 6[n] [4], as compared to similar benzene mesogens [24]. Also in series 7–11 the introduction of the connecting oxygen atom gives a larger increase in mesophase stability by an average of 6 ± 2 K for the 12-vertex p-carborane derivatives than for their benzene analogues [5]. However, in series 12 and 13 the effect of incorporation of the O atom as the connecting group is practically the same for all ring systems [15].
![[1860-5397-5-83-2]](/bjoc/content/figures/1860-5397-5-83-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The molecular structures of derivatives 1–20.
Figure 2: The molecular structures of derivatives 1–20.
A recently developed series of isostructural mesogens allows to analyze the effect of the replacement of an alkoxy in 14[6] with an ester group in 15[6]. The CH2O→OOC exchange dramatically destabilized the nematic phase for the 10- and 12-vertex p-carborane derivatives, while a much smaller effect was observed for the carbocycles [25]. Series 14 and 15 [25] and also diesters 17 [2] provide an opportunity for further investigation of this interesting phenomenon. Therefore, we focused on series 14–16 to investigate the CH2O, COO, OOC connecting groups, and also on series 17–20 to study the CH2O, CH2CH2, COO, OOC groups.
Here we describe the preparation of an isostructural series of diesters 16[6] and 18B, and also tetraesters 19 and 20, and a detailed comparative analysis focusing on the impact of the connecting group on mesogenic properties. The analysis is aided by molecular modeling of the pertinent molecular fragments. In addition, we report two homologues of 16A[6], diesters 16A[5] and 16A[7].
Results
Synthesis
Diesters 16[n] were prepared from diphenols 21 and appropriate carboxylic acid chlorides in the presence of a base as shown in Scheme 1. The requisite diphenols 21 were obtained in nearly quantitative yields by treating the corresponding dimethoxy derivatives 14[0] with BBr3. This procedure represents a significant improvement to the original preparation of 1,12-bis(4-hydroxyphenyl)-p-carborane (21A) from 14A[0] [26] and 1,4-bis(4-hydroxyphenyl)benzene (21D) [27]. The preparation of diphenol 21C will be described elsewhere [25]. The dimethoxy carborane derivative 14A[0] was obtained using the Wade’s arylation procedure [28] of p-carborane (A) with 4-iodoanisole as described before [26]. The 10-vertex analogue 14B[0] was prepared in a similar way and will be described elsewhere [25]. 4,4”-Dimethoxyterphenyl 14D[0] was prepared in 84% yield from 1,4-dibromobenzene and (4-methoxyphenyl)boronic acid using the Suzuki coupling procedure [29]. This method is comparable to other Pd(0)-assisted methods for the synthesis of 14D[0] [30-32].
![[1860-5397-5-83-i1]](/bjoc/content/inline/1860-5397-5-83-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The 10-vertex p-carborane diester 18B was obtained from the corresponding dicarboxylic acid 22B [33] and 4-pentylphenol (Scheme 2) according to a recently described procedure [2]. The two series of tetraesters 19 and 20 were prepared from the appropriate dicarboxylic acid 22 and phenols 23 and 24, respectively. p-Carborane-1,12-dicarboxylic acid 22A and terephthalic acid (22D) were converted to the corresponding acid chlorides using PCl5 and then reacted with phenols in the presence of a base. The previously described method [2] for the preparation of esters of bicyclo[2.2.2]octane-1,4-dicarboxylic acid (22C) was unsuccessful and the desired esters 19C and 20C were obtained from the diacid and appropriate phenol using the classical Mitsunobu procedure [34]. A similar procedure was used for the preparation of tetraester 20B, while 19B was prepared more efficiently using the acid chloride method. Ester 19D has been reported in the literature [35].
![[1860-5397-5-83-i2]](/bjoc/content/inline/1860-5397-5-83-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Phenol 24 was prepared by acylation of 4-benzyloxyphenol with butyryl chloride followed by removal of the protective benzyl group under reductive conditions as described in the literature [36] (Scheme 3).
![[1860-5397-5-83-i3]](/bjoc/content/inline/1860-5397-5-83-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Mesogenic properties
Transition temperatures and enthalpies of the newly prepared compounds were determined by differential scanning calorimetry (DSC). The phase types were assigned by comparison of microscopic textures observed in polarized light with those published for reference compounds [37-39]. Results for these and also their structural analogues are shown in Table 1 and Table 2.
Table 1: Transition temperatures (°C) for selected liquid crystals.a
| * | A | B | C | D | |
|---|---|---|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-5-83-i4.svg?max-width=637&scale=1.18182)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-83-i5.svg?max-width=637&scale=1.18182)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-5-83-i6.svg?max-width=637&scale=1.18182)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-5-83-i7.svg?max-width=637&scale=1.18182)
|
||
| X |
![[Graphic 5]](/bjoc/content/inline/1860-5397-5-83-i8.svg?max-width=637&scale=1.0)
|
||||
| 14[6] | –CH2O–(Ph) | Cr 96 N 98 Ib | Cr 73 N 105 Ib | Cr 98 SmB 161 SmA 179 Ib | Crc 182 SmF 218 SmI 219 SmC 232 SmA 235 Ib |
| 15[6] | –OOC–(Ph) | Cr 112 (N 31) Ib | Cr 65 (N 11) Ib | Crd 114 SmA 148 Ib | Cr 134 SmC 143 SmA 183 Ib |
| 16[6] | –COO–(Ph) | Cr 108 N 132 I | Cre 102 N 136 I | Crf 102 X 205 N 207 I | Cr 66 X 96 SmF 226 SmI 232 SmCg 250 SmA 251 I |
![[Graphic 6]](/bjoc/content/inline/1860-5397-5-83-i9.svg?max-width=637&scale=1.0)
|
|||||
| 17 | –CH2O–(Ph) | Cr 137 N 182.6 Ih | Cri 111 N 183.4 Ih | Cr 112 N 229.5 Ih | Cr 189 N 235 Ij |
| 18 | –CH2CH2–(Ph) | Cr 106 N 118 Ik | Cr 85 N 110 I | Cr 98 N 173 Il | Cr 155 N 181 Im |
| 19 | –OOC–(Ph) | Cr 203 (N 139) Ig | Cr 160 (N 128) I | Cr 121 N 195 I | Cr 130 SmA 207 N 221 In |
| 20 | –COO–(Ph) | Cr 133 N 230 I | Cr 120 N 234 I | Cr 133 N 275 I | Cr 230 N 287 I |
aObtained on heating; Cr: crystal, Sm: smectic, N: nematic, I: isotropic, X: unidentified phase. Transition enthalpies for new compounds are listed in the SI. bRef. [25]. cCr – Cr transition at 108 °C. dCr – Cr transition at 100 °C. eCr – Cr transition at 73 °C (14.9 kJ/mol). fCr – Cr transition at 33 °C (11.6 kJ/mol). gOptical determination obtained on cooling .hRef. [2]. iCr – Cr transition at 70 °C. jRef. [40]. kPreviously reported Cr 104 N 114 I, ref. [21]. lRef. [41]. mRef. [42]. nRef. [35].
Table 2: Transition temperatures (°C) for 16A[n].a
![[Graphic 7]](/bjoc/content/inline/1860-5397-5-83-i10.svg?max-width=637&scale=1.0)
|
|
| n | Transition temperatures |
|---|---|
| 5 | Cr1 66 Cr2 120 N 155 I |
| 6 | Cr 108 N 132 I |
| 7 | Cr1 76 Cr2 92 N 124 I |
aObtained on heating; Cr: crystal, N: nematic, I: isotropic. Transition enthalpies are listed in the SI.
All p-carborane derivatives in series 14–20 exhibit exclusively the nematic phase. Similar nematic behavior is observed for carbocycles in series 17–20 with the exception of 19D, which exhibits a SmA phase in addition to a N phase. In contrast, most carbocyclic derivatives in series 14[6]–16[6] display only smectic and soft crystalline polymorphs. The bicyclo[2.2.2]octane derivative 16C[6] is the only exception and exhibits a narrow range nematic phase above a soft crystalline phase designated as L or E on the basis of viscosity, ability to supercool, and optical textures. In general, bicyclo[2.2.2]octane derivatives 14C[6]–16C[6] exhibit orthogonal phases (SmA and SmB), while the terphenyl analogues display a rich smectic polymorphism involving mainly tilted phases. The terphenyl derivatives 14D[6] and 16D[6] exhibit the most interesting polymorphism in the series with 4 smectic phases and possibly a soft crystalline modification such as a G phase below the SmF phase in the latter. A DSC trace for 16D[6] is shown in Figure 3, and representative textures of its mesophases are presented in Figure 4. The tilted phases in both terphenyl compounds were identified by the appearance and subsequent characteristic changes of the Schlieren textures in the homeotropic regions of the SmA phase upon cooling.
![[1860-5397-5-83-3]](/bjoc/content/figures/1860-5397-5-83-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Partial DSC trace for 16D[6]. Heating rate 5 K/min.
Figure 3: Partial DSC trace for 16D[6]. Heating rate 5 K/min.
![[1860-5397-5-83-4]](/bjoc/content/figures/1860-5397-5-83-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Optical textures of 16D[6] obtained for the same region of the sample upon cooling: (a) SmA growing from isotropic (251 °C), (b) focal-conic texture of SmC (242 °C), (c) SmI (229 °C), and (d) broken focal-conic texture of SmF (211 °C). Magnification × 60.
Figure 4: Optical textures of 16D[6] obtained for the same region of the sample upon cooling: (a) SmA growing...
In general, the order of phase stability for all five series follows A ~ B < C < D. Derivatives of both p-carboranes A and B exhibit similar stability of the nematic phase, with the exception of 15[6] and 19 for which the monotropic nematic phase of the 10-vertex carborane derivatives is significantly less stable (<20 K) than that of the 12-vertex analogues.
Analysis of three homologues 16A[n] demonstrated the decreasing stability of the nematic phase with increasing chain length from TNI of 155 °C for n = 5 to 124 °C for n = 7 (Table 2). Investigation of the 4,4″-dimethoxyterphenyl 14D[0] revealed a high temperature nematic phase (Cr 277 N 295 I), which is in disagreement with the original literature report [43].
Comparative Analysis
Mesogenic properties of structurally analogous pairs were compared, and the results are presented in Figure 5 and Figure 6.
![[1860-5397-5-83-5]](/bjoc/content/figures/1860-5397-5-83-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The change in the clearing temperature ΔTc upon substitution –OCH2– → –CH2CH2– in selected pairs of compounds. The lines are guides for the eye.
Figure 5: The change in the clearing temperature ΔTc upon substitution –OCH2– → –CH2CH2– in selected pairs of...
![[1860-5397-5-83-6]](/bjoc/content/figures/1860-5397-5-83-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The change in the clearing temperature ΔTc upon replacing of the –OCH2– connecting group with another in selected pairs of compounds. The lines are guides for the eye.
Figure 6: The change in the clearing temperature ΔTc upon replacing of the –OCH2– connecting group with anoth...
The –OCH2– → –CH2CH2– substitution
A comparison of TNI for compounds in series 17 [2] versus their isostructural analogues 18, in which the linking oxygen atom is replaced with –CH2–, demonstrates that the presence of the oxygen atom increases the phase stability by about 55 K (or 27 K per alkoxyphenyl group) for the carbocyclic compounds. In contrast, the difference in TNI is larger by 10 K for p-carborane A and 18 K for p-carborane B (Figure 5 and Figure 6). These results are consistent with earlier findings for pairs 1/2, 3/4, and 5/6 (Figure 5) in which a particularly large impact of the –OCH2– → –CH2CH2– substitution on TNI is observed for the rigid biphenyl derivatives 1–4.
The –OCH2– → –OOC– and –OCH2– →–COO– substitution
Data in Table 1 demonstrate that the replacement of the heptyloxy group with heptanoyloxy in 14[16]/16[6] and butoxy with butanoyloxy in 17/20 results in an increase of the TNI by about 30 K and 45 K, respectively, for all structural units A–D. The only exception is the pair 14D[6]/16D[6] for which the change in TNI is only 16 K. The larger change of TNI for pairs 17/20 than for 14[6]/16[6] is consistent with attenuation of the substitution effect by the shorter alkyl chain in the latter (–C3H7 vs –C6H13).
In contrast, replacement of the oxymethylene linking group with a carboxy group of reversed orientation relative to the core (pairs 14[6]/15[6] and 17/19) leads to significant destabilization of the mesophase, and the magnitude of the effect markedly depends on the nature of the central structural element (Figure 6). Thus, for derivatives of carbocycles C and D, Tc decreases less than 55 K for 14[6]/15[6] and less than 40 K for 17/19, while for the p-carboranes the decrease is larger, reaching a value of 94 K for the pair 14B[6]/15B[6].
Data in Table 1 also allow for assessment of the impact of the orientation of the connecting carboxyl group on TNI as a function of the central structural element. Thus, in pairs 16[6]/15[6] and 20/19, the change of carbonyloxy to oxycarbonyl leads to a marked phase destabilization for all structural elements A–D. Consistent with our previous analysis, the effect is much stronger for p-carboranes (>90 K) than for carbocycles (<80 K) with the typical order: D, C < A < B.
Molecular Modeling
For a better understanding of the terminal substituent’s impact on the conformational ground state of the molecules, four benzene derivative models, 25–28, were optimized at the B3LYP/6-31G(d) level of theory, and their equilibrium geometries are presented in Figure 7. Results show that the replacement of the oxygen atom with a –CH2– group reorients the terminal chain from co-planar, in the conformational ground state of ethoxybenzene (25), to the orthogonal position relative to the benzene ring plane in propylbenzene (26). Replacement of the –CH2O– fragment with the –COO– group leads to an increase of angle θ between the ring and substituent planes to about 50° in phenyl acetate (27). Reversing the connectivity of the ester group (–COO– → –OOC–) results in return to the co-planar orientation of the substituent in benzoate 28. The computational results are consistent with experimental findings for anisole [44] and ethylbenzene [45], and solid-state structures for compounds containing fragments 25–28 [46].
![[1860-5397-5-83-7]](/bjoc/content/figures/1860-5397-5-83-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Equilibrium ground state geometries (B3LYP/6-31G(d)) for benzene derivatives: ethoxybenzene (25), propylbenzene (26), phenyl acetate (27), and methyl benzoate (28) and pertinent molecular parameters: dihedral angle θ, dipole moment μ, and quadrupole moment tensor QXX perpendicular to the ring plane.
Figure 7: Equilibrium ground state geometries (B3LYP/6-31G(d)) for benzene derivatives: ethoxybenzene (25), p...
Overall, the interplanar angle θ between the ring and substituent increases in the series 25, 28 < 27 < 26 or –OR, –C(O)OR < –OOCR < –CH2R.
Further analysis of the computational results demonstrates that molecular dipole moment μ increases in the following order: 26 < 25 < 27 < 28. The quadrupole moment tensor Qxx perpendicular to the benzene ring is larger for the esters than for alkyl or alkoxy derivatives.
Discussion and Conclusion
Results presented in Table 1 are in agreement with general trends [47,48] and demonstrate that the replacement of –OCH2– with –OOC– increases TNI, while replacement with –CH2CH2– or –COO– decreases TNI. Overall, the effectiveness of the connecting group in mesophase stabilization follows the order: –OOC(Alk) > –OCH2(Alk) > –COO(Alk) > –CH2CH2(Alk). The magnitude of the effect for the –OCH2– → –OOC– replacement is practically independent of the central structural element A–D. In contrast, the decrease in TNI upon substitution of –OCH2– with –CH2CH2– or –COO– is stronger for p-carborane derivatives (A and B) than for their carbocyclic analogues. This effect is observed for compounds in which the p-carborane is connected directly to the substituted benzene ring (1–4, 14–16) or through a spacer (5, 6, 17–20).
The origin of the observed relative effectiveness of the connecting groups (–OOC(Alk) > –OCH2(Alk) > –COO(Alk) > –CH2CH2(Alk)) is unclear at the moment. In general, the phase stability is related to packing fraction, and for more compact anisometric molecules (high packing fraction) the clearing temperature is higher [49]. Thus, it can be expected that compounds with substituents preferring coplanar orientation with the aryl ring (–OR and –COOR) would exhibit higher mesophase stability than those with non-coplanar orientation (–CH2R and –OOCR). While this simple steric argument is consistent with data for pairs –CH2R/–OR, the effect of orientation of the carboxy group on mesophase stability is opposite. Therefore, steric arguments alone cannot explain the observed trend for these analogues.
Electronic effects also cannot sufficiently explain the observed trend in mesophase stability and the poor performance of the carboxyl group. Thus, the observed trend is inconsistent with the order of dipole moments calculated for the relevant molecular fragments 25–28 (Figure 7). According to the computational results, esters 27 and 28 have similar, and also the largest molecular dipole and quadrupole moments. Therefore, compounds containing these fragments would be expected to exhibit both similar and high mesophase stability. The data show otherwise and a large difference in TC is observed for the isomeric esters (e.g. for 19B/20B ΔTNI = 80 K; for other examples see LiqCryst database [50]).
The origin of the observed excessive mesophase destabilization in p-carborane derivatives by the –COO(Alk) and –CH2CH2(Alk) substituents is even more puzzling. Data in Table 1 show that mesophase of p-carborane derivatives containing electron rich benzene rings (with the –OR and –OOCR substituents) is excessively stabilized relative to those containing either weakly donating (–CH2CH2R) or electron-withdrawing (–COOR) substituents. This suggests that intermolecular quadrupolar interactions between p-carborane and benzene ring may be responsible for the observed phase stabilization. Support for this hypothesis is provided by the finding that the connecting group affects bulk properties whether p-carborane is connected to the benzene ring directly or through a spacer. The observed larger effect of the –CH2CH2– → –OCH2– replacement in pairs 1A/2A and 3A/4A as compared to 17A/18A suggests a role for the molecular dipole moment in phase stabilization. Since p-carboranes are moderately electron withdrawing substituents, the alkoxy derivatives have a larger dipole moment than the alkyl derivatives [16]. Alternatively, the effect can be due to higher rigidity of 1-4, which attenuates the effect as compared to the more conformationally flexible diesters.
Overall, the analysis cannot distinguish one particular factor responsible for the impact of structural elements (A–D) on phase stabilization. Instead, a combination of conformational properties of structural elements A–D and substituents, their relative sizes [51], and electronic properties of the benzene ring bearing the substituent dictate mesogenic properties.
The present report concentrates on the systematic variation of the connecting group between the alkyl and phenyl ring, and its effect on phase stability. For completeness, we also mention one example of variation of the carborane–alkyl connecting group, and its impact on Tc. Thus, a replacement of –CH2CH2– → –C≡C– destabilized the TNI by over 150 K in bi-carborane derivatives, while a similar transformation in the biphenyl analogue leads to an increase of the clearing temperature [1,7]. This dramatic effect has been attributed to conformational properties of molecules in the condensed phase.
A more complete understanding of the impact of structural modification on bulk properties will emerge through further research on structure-property relationships and studying of other examples of structurally similar mesogens containing the four ring systems A–D.
Experimental
Optical microscopy and phase identification were performed using a PZO “Biolar” polarized microscope equipped with a HCS400 Instec hot stage. Thermal analysis was obtained using a TA Instruments 2920 DSC. Transition temperatures (onset) and enthalpies were obtained using small samples (1–2 mg) and a heating rate of 5 K min−1 under a flow of nitrogen gas. For DSC and microscopic analyses, each compound was additionally purified by dissolving in CH2Cl2, filtering to remove particles, evaporating and recrystallization typically from hexanes or toluene/heptane mixture. The resulting crystals were dried in vacuum overnight at ambient temperature. 10- and 12-vertex p-carboranes (A and B) were purchased from Katchem s. r. o. (Prague, Czech Republic).
Supporting Information
Synthetic procedures for compounds 14D[0], 16[n], 18B, 19, 20, 24, and analytical details are provided.
| Supporting Information File 1: General methods and synthetic procedures | ||
| Format: PDF | Size: 70.8 KB | Download |
Acknowledgments
We are grateful to Mr. Bryan Ringstrand for the photomicrographs of 16D[6]. Financial support for this work was received from the National Science Foundation (DMR-0606317 and DMR-0907542) and from the Ministry of Education, Culture, Sports, Science and Technology, Japan (Grant-in-Aid for Scientific Research (B) No. 13470468).
References
-
Kaszynski, P.; Pakhomov, S.; Tesh, K. F.; Young, V. G., Jr. Inorg. Chem. 2001, 40, 6622–6631. doi:10.1021/ic010663x
Return to citation in text: [1] [2] [3] -
Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Nagamine, T.; Januszko, A.; Ohta, K.; Kaszynski, P.; Endo, Y. Liq. Cryst. 2005, 32, 985–995. doi:10.1080/02678290500291756
Return to citation in text: [1] -
Nagamine, T.; Januszko, A.; Kaszynski, P.; Ohta, K.; Endo, Y. J. Mater. Chem. 2006, 16, 3836–3843. doi:10.1039/b608012j
Return to citation in text: [1] [2] -
Nagamine, T.; Januszko, A.; Ohta, K.; Kaszynski, P.; Endo, Y. Liq. Cryst. 2008, 35, 865–884. doi:10.1080/02678290802245450
Return to citation in text: [1] [2] [3] -
Ohta, K.; Januszko, A.; Kaszynski, P.; Nagamine, T.; Sasnouski, G.; Endo, Y. Liq. Cryst. 2004, 31, 671–682. doi:10.1080/02678290410001670584
Return to citation in text: [1] -
Piecek, W.; Kaufman, J. M.; Kaszynski, P. Liq. Cryst. 2003, 30, 39–48. doi:10.1080/0267829021000039669
Return to citation in text: [1] [2] -
Ringstrand, B.; Vroman, J.; Jensen, D.; Januszko, A.; Kaszynski, P.; Dziaduszek, J.; Drzewinski, W. Liq. Cryst. 2005, 32, 1061–1070. doi:10.1080/02678290500291699
Return to citation in text: [1] [2] -
Douglass, A. G.; Czuprynski, K.; Mierzwa, M.; Kaszynski, P. Chem. Mater. 1998, 10, 2399–2402. doi:10.1021/cm980089w
Return to citation in text: [1] -
Douglass, A. G.; Mierzwa, M.; Kaszynski, P. Liquid Crystals Containing p-Carborane. In Liquid Crystals: Chemistry and Structure; Tykarska, M.; Dabrowski, R.; Zielinski, J., Eds.; Zakopane, 1998; Vol. 3319, pp 59–62.
Return to citation in text: [1] -
Czuprynski, K.; Douglass, A. G.; Kaszynski, P.; Drzewinski, W. Liq. Cryst. 1999, 26, 261–269. doi:10.1080/026782999205407
Return to citation in text: [1] -
Douglass, A. G.; Both, B.; Kaszynski, P. J. Mater. Chem. 1999, 9, 683–686. doi:10.1039/a807596d
Return to citation in text: [1] -
Czuprynski, K.; Kaszynski, P. Liq. Cryst. 1999, 26, 775–778. doi:10.1080/026782999204877
Return to citation in text: [1] -
Douglass, A. G.; Czuprynski, K.; Mierzwa, M.; Kaszynski, P. J. Mater. Chem. 1998, 8, 2391–2398. doi:10.1039/a804322a
Return to citation in text: [1] -
Januszko, A.; Kaszynski, P.; Wand, M. D.; More, K. M.; Pakhomov, S.; O’Neill, M. J. Mater. Chem. 2004, 14, 1544–1553. doi:10.1039/b311140g
Return to citation in text: [1] [2] -
Januszko, A.; Glab, K. L.; Kaszynski, P.; Patel, K.; Lewis, R. A.; Mehl, G. H.; Wand, M. D. J. Mater. Chem. 2006, 16, 3183–3192. doi:10.1039/b600068a
Return to citation in text: [1] [2] [3] -
Januszko, A.; Kaszynski, P.; Drzewinski, W. J. Mater. Chem. 2006, 16, 452–461. doi:10.1039/b512319d
Return to citation in text: [1] [2] -
Januszko, A.; Kaszynski, P. Liq. Cryst. 2008, 35, 705–710. doi:10.1080/02678290802120281
Return to citation in text: [1] [2] -
Januszko, A.; Glab, K. L.; Kaszynski, P. Liq. Cryst. 2008, 35, 549–553. doi:10.1080/02678290802015713
Return to citation in text: [1] [2] -
Jasinski, M.; Jankowiak, A.; Januszko, A.; Bremer, M.; Pauluth, D.; Kaszynski, P. Liq. Cryst. 2008, 35, 343–350. doi:10.1080/02678290701817318
Return to citation in text: [1] [2] -
Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705
Return to citation in text: [1] [2] -
Kaszynski, P.; Douglass, A. G. J. Organomet. Chem. 1999, 581, 28–38. doi:10.1016/S0022-328X(99)00088-1
Return to citation in text: [1] -
Kaszynski, P. closo-Boranes as π Structural Elements for Advanced Anisotropic Materials. In Anisotropic Organic Materials-Approaches to Polar Order; Glaser, R.; Kaszynski, P., Eds.; ACS Symposium Series: Washington, D.C., 2001; Vol. 798, pp 68–82.
and references therein.
Return to citation in text: [1] [2] -
Villiger, A.; Leenhouts, F. Mol. Cryst. Liq. Cryst. 1991, 209, 297–307. doi:10.1080/00268949108036205
Return to citation in text: [1] -
Kaszynski, P.; Kulikiewicz, K. K.; Januszko, A.; Douglass, A. G.; Tilford, R. W.; Pakhomov, S.; Patel, M. K.; Ke, Y.; Radziszewski, G. J.; Young, V. G., Jr.
submitted.
Return to citation in text: [1] [2] [3] [4] [5] -
Fox, M. A.; MacBride, J. A. H.; Peace, R. J.; Wade, K. J. Chem. Soc., Dalton Trans. 1998, 401–412. doi:10.1039/a707154j
Return to citation in text: [1] [2] -
Price, C. C.; Mueller, G. P. J. Am. Chem. Soc. 1944, 66, 632–634. doi:10.1021/ja01232a038
Return to citation in text: [1] -
Coult, R.; Fox, M. A.; Gill, W. R.; Herbertson, P. L.; McBride, J. A. H.; Wade, K. J. Organomet. Chem. 1993, 462, 19–29. doi:10.1016/0022-328X(93)83337-U
Return to citation in text: [1] -
Miyaura, N.; Yanagi, Y.; Suzuki, A. Synth. Commun. 1981, 11, 513–519. doi:10.1080/00397918108063618
Return to citation in text: [1] -
Chaumeil, H.; Le Drian, C.; Defoin, A. Synthesis 2002, 757–760. doi:10.1055/s-2002-25773
Return to citation in text: [1] -
Sinclair, D. J.; Sherburn, M. S. J. Org. Chem. 2005, 70, 3730–3733. doi:10.1021/jo050105q
Return to citation in text: [1] -
Tao, X.; Zhao, Y.; Shen, D. Synlett 2004, 359–361. doi:10.1055/s-2003-44992
Return to citation in text: [1] -
Garrett, P. M.; Smart, J. C.; Hawthorne, M. F. J. Am. Chem. Soc. 1969, 91, 4707–4709. doi:10.1021/ja01045a021
Return to citation in text: [1] -
Mitsunobu, O. Synthesis 1981, 1–28. doi:10.1055/s-1981-29317
Return to citation in text: [1] -
Leblanc, J. P.; Tessier, M.; Judas, D.; Friedrich, C.; Noël, C.; Maréchal, E. Macromolecules 1993, 26, 4391–4399. doi:10.1021/ma00069a001
Return to citation in text: [1] [2] -
Neubert, M. E.; Wildman, P. J.; Zawaski, M. J.; Hanlon, C. A.; Benyo, T. L.; De Vries, A. Mol. Cryst. Liq. Cryst. 1987, 145, 111–158. doi:10.1080/00268948708080217
Return to citation in text: [1] -
Demus, D.; Richter, L. Textures of Liquid Crystals, 2nd ed.; VEB Deutscher Verlag für Grundstoffindustrie: Leipzig, 1980.
Return to citation in text: [1] -
Gray, G. W.; Goodby, J. W. G. Smectic Liquid Crystals-Textures and Structures; Leonard Hill: Philadelphia, 1984.
Return to citation in text: [1] -
Dierking, I. Smectic Liquid Crystals Textures and Structures; Wiley-VCH: Weinheim, 2003. doi:10.1002/3527602054
Return to citation in text: [1] -
Kelker, H.; Scheurle, B. J. Phys. (Paris) 1969, 30-C4, 104–108. doi:10.1051/jphyscol:1969425
Return to citation in text: [1] -
Compound ID # 37494 in LiqCryst 4.6 database.
Return to citation in text: [1] -
Neubert, M. E.; Stahl, M. E.; Cline, R. E. Mol. Cryst. Liq. Cryst. 1982, 89, 93–117. doi:10.1080/00268948208074472
Return to citation in text: [1] -
Compound ID # 21727 in LiqCryst 4.6 database.
Return to citation in text: [1] -
Onda, M.; Toda, A.; Mori, S.; Yamaguchi, I. J. Mol. Struct. 1986, 144, 47–51. doi:10.1016/0022-2860(86)80166-1
Return to citation in text: [1] -
Scharfenberg, P.; Rozsondai, B.; Hargittai, I. Z. Naturforsch., A 1980, 35, 431–436.
Return to citation in text: [1] -
Haase, W.; Athanassopoulou, M. A. Crystal Structures of LC Mesogens. In Structure and Bonding; Mingos, D. M. P., Ed.; Springer: Berlin, 1999; Vol. 94, pp 139–197.
Return to citation in text: [1] -
Toyne, K. J. Liquid Crystal Behavior in Relation to Molecular Structure. In Thermotropic Liquid Crystals; Gray, G. W., Ed.; Wiley: New York, 1987; pp 28–63.
Return to citation in text: [1] -
Demus, D. Chemical Structure and Mesogenic Properties. In Handbook of Liquid Crystals; Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: New York, 1998; Vol. 1, pp 133–187.
Return to citation in text: [1] -
Demus, D.; Hauser, A. Molecular structure and thermodynamic properties of nematic liquid crystals. In Selected Topics In Liquid Crystal Research; Koswig, H.-D., Ed.; Akademie-Verlag: Berlin, 1990; pp 19–44.
Return to citation in text: [1] -
Vill, V. LiqCryst 4.6 database (LCI Publisher GmbH, Hamburg, Germany, www.lci-publisher.com) and references therein.
Return to citation in text: [1] -
Effective diameter d for the four structural elements: A, d = 7.43 Å; B, d = 7.17 Å; C, d = 6.72 Å; D, d = 5.03 Å. For details see footnote 33 in reference 16.
Return to citation in text: [1]
| 36. | Neubert, M. E.; Wildman, P. J.; Zawaski, M. J.; Hanlon, C. A.; Benyo, T. L.; De Vries, A. Mol. Cryst. Liq. Cryst. 1987, 145, 111–158. doi:10.1080/00268948708080217 |
| 37. | Demus, D.; Richter, L. Textures of Liquid Crystals, 2nd ed.; VEB Deutscher Verlag für Grundstoffindustrie: Leipzig, 1980. |
| 38. | Gray, G. W.; Goodby, J. W. G. Smectic Liquid Crystals-Textures and Structures; Leonard Hill: Philadelphia, 1984. |
| 39. | Dierking, I. Smectic Liquid Crystals Textures and Structures; Wiley-VCH: Weinheim, 2003. doi:10.1002/3527602054 |
| 25. |
Kaszynski, P.; Kulikiewicz, K. K.; Januszko, A.; Douglass, A. G.; Tilford, R. W.; Pakhomov, S.; Patel, M. K.; Ke, Y.; Radziszewski, G. J.; Young, V. G., Jr.
submitted. |
| 1. | Kaszynski, P.; Pakhomov, S.; Tesh, K. F.; Young, V. G., Jr. Inorg. Chem. 2001, 40, 6622–6631. doi:10.1021/ic010663x |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 3. | Nagamine, T.; Januszko, A.; Ohta, K.; Kaszynski, P.; Endo, Y. Liq. Cryst. 2005, 32, 985–995. doi:10.1080/02678290500291756 |
| 4. | Nagamine, T.; Januszko, A.; Kaszynski, P.; Ohta, K.; Endo, Y. J. Mater. Chem. 2006, 16, 3836–3843. doi:10.1039/b608012j |
| 5. | Nagamine, T.; Januszko, A.; Ohta, K.; Kaszynski, P.; Endo, Y. Liq. Cryst. 2008, 35, 865–884. doi:10.1080/02678290802245450 |
| 6. | Ohta, K.; Januszko, A.; Kaszynski, P.; Nagamine, T.; Sasnouski, G.; Endo, Y. Liq. Cryst. 2004, 31, 671–682. doi:10.1080/02678290410001670584 |
| 7. | Piecek, W.; Kaufman, J. M.; Kaszynski, P. Liq. Cryst. 2003, 30, 39–48. doi:10.1080/0267829021000039669 |
| 8. | Ringstrand, B.; Vroman, J.; Jensen, D.; Januszko, A.; Kaszynski, P.; Dziaduszek, J.; Drzewinski, W. Liq. Cryst. 2005, 32, 1061–1070. doi:10.1080/02678290500291699 |
| 9. | Douglass, A. G.; Czuprynski, K.; Mierzwa, M.; Kaszynski, P. Chem. Mater. 1998, 10, 2399–2402. doi:10.1021/cm980089w |
| 10. | Douglass, A. G.; Mierzwa, M.; Kaszynski, P. Liquid Crystals Containing p-Carborane. In Liquid Crystals: Chemistry and Structure; Tykarska, M.; Dabrowski, R.; Zielinski, J., Eds.; Zakopane, 1998; Vol. 3319, pp 59–62. |
| 11. | Czuprynski, K.; Douglass, A. G.; Kaszynski, P.; Drzewinski, W. Liq. Cryst. 1999, 26, 261–269. doi:10.1080/026782999205407 |
| 12. | Douglass, A. G.; Both, B.; Kaszynski, P. J. Mater. Chem. 1999, 9, 683–686. doi:10.1039/a807596d |
| 13. | Czuprynski, K.; Kaszynski, P. Liq. Cryst. 1999, 26, 775–778. doi:10.1080/026782999204877 |
| 14. | Douglass, A. G.; Czuprynski, K.; Mierzwa, M.; Kaszynski, P. J. Mater. Chem. 1998, 8, 2391–2398. doi:10.1039/a804322a |
| 15. | Januszko, A.; Kaszynski, P.; Wand, M. D.; More, K. M.; Pakhomov, S.; O’Neill, M. J. Mater. Chem. 2004, 14, 1544–1553. doi:10.1039/b311140g |
| 16. | Januszko, A.; Glab, K. L.; Kaszynski, P.; Patel, K.; Lewis, R. A.; Mehl, G. H.; Wand, M. D. J. Mater. Chem. 2006, 16, 3183–3192. doi:10.1039/b600068a |
| 17. | Januszko, A.; Kaszynski, P.; Drzewinski, W. J. Mater. Chem. 2006, 16, 452–461. doi:10.1039/b512319d |
| 18. | Januszko, A.; Kaszynski, P. Liq. Cryst. 2008, 35, 705–710. doi:10.1080/02678290802120281 |
| 19. | Januszko, A.; Glab, K. L.; Kaszynski, P. Liq. Cryst. 2008, 35, 549–553. doi:10.1080/02678290802015713 |
| 20. | Jasinski, M.; Jankowiak, A.; Januszko, A.; Bremer, M.; Pauluth, D.; Kaszynski, P. Liq. Cryst. 2008, 35, 343–350. doi:10.1080/02678290701817318 |
| 21. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 22. | Kaszynski, P.; Douglass, A. G. J. Organomet. Chem. 1999, 581, 28–38. doi:10.1016/S0022-328X(99)00088-1 |
| 23. |
Kaszynski, P. closo-Boranes as π Structural Elements for Advanced Anisotropic Materials. In Anisotropic Organic Materials-Approaches to Polar Order; Glaser, R.; Kaszynski, P., Eds.; ACS Symposium Series: Washington, D.C., 2001; Vol. 798, pp 68–82.
and references therein. |
| 18. | Januszko, A.; Kaszynski, P. Liq. Cryst. 2008, 35, 705–710. doi:10.1080/02678290802120281 |
| 19. | Januszko, A.; Glab, K. L.; Kaszynski, P. Liq. Cryst. 2008, 35, 549–553. doi:10.1080/02678290802015713 |
| 26. | Fox, M. A.; MacBride, J. A. H.; Peace, R. J.; Wade, K. J. Chem. Soc., Dalton Trans. 1998, 401–412. doi:10.1039/a707154j |
| 5. | Nagamine, T.; Januszko, A.; Ohta, K.; Kaszynski, P.; Endo, Y. Liq. Cryst. 2008, 35, 865–884. doi:10.1080/02678290802245450 |
| 27. | Price, C. C.; Mueller, G. P. J. Am. Chem. Soc. 1944, 66, 632–634. doi:10.1021/ja01232a038 |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 1. | Kaszynski, P.; Pakhomov, S.; Tesh, K. F.; Young, V. G., Jr. Inorg. Chem. 2001, 40, 6622–6631. doi:10.1021/ic010663x |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 25. |
Kaszynski, P.; Kulikiewicz, K. K.; Januszko, A.; Douglass, A. G.; Tilford, R. W.; Pakhomov, S.; Patel, M. K.; Ke, Y.; Radziszewski, G. J.; Young, V. G., Jr.
submitted. |
| 42. | Neubert, M. E.; Stahl, M. E.; Cline, R. E. Mol. Cryst. Liq. Cryst. 1982, 89, 93–117. doi:10.1080/00268948208074472 |
| 23. |
Kaszynski, P. closo-Boranes as π Structural Elements for Advanced Anisotropic Materials. In Anisotropic Organic Materials-Approaches to Polar Order; Glaser, R.; Kaszynski, P., Eds.; ACS Symposium Series: Washington, D.C., 2001; Vol. 798, pp 68–82.
and references therein. |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 35. | Leblanc, J. P.; Tessier, M.; Judas, D.; Friedrich, C.; Noël, C.; Maréchal, E. Macromolecules 1993, 26, 4391–4399. doi:10.1021/ma00069a001 |
| 24. | Villiger, A.; Leenhouts, F. Mol. Cryst. Liq. Cryst. 1991, 209, 297–307. doi:10.1080/00268949108036205 |
| 15. | Januszko, A.; Kaszynski, P.; Wand, M. D.; More, K. M.; Pakhomov, S.; O’Neill, M. J. Mater. Chem. 2004, 14, 1544–1553. doi:10.1039/b311140g |
| 21. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 4. | Nagamine, T.; Januszko, A.; Kaszynski, P.; Ohta, K.; Endo, Y. J. Mater. Chem. 2006, 16, 3836–3843. doi:10.1039/b608012j |
| 25. |
Kaszynski, P.; Kulikiewicz, K. K.; Januszko, A.; Douglass, A. G.; Tilford, R. W.; Pakhomov, S.; Patel, M. K.; Ke, Y.; Radziszewski, G. J.; Young, V. G., Jr.
submitted. |
| 17. | Januszko, A.; Kaszynski, P.; Drzewinski, W. J. Mater. Chem. 2006, 16, 452–461. doi:10.1039/b512319d |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 8. | Ringstrand, B.; Vroman, J.; Jensen, D.; Januszko, A.; Kaszynski, P.; Dziaduszek, J.; Drzewinski, W. Liq. Cryst. 2005, 32, 1061–1070. doi:10.1080/02678290500291699 |
| 16. | Januszko, A.; Glab, K. L.; Kaszynski, P.; Patel, K.; Lewis, R. A.; Mehl, G. H.; Wand, M. D. J. Mater. Chem. 2006, 16, 3183–3192. doi:10.1039/b600068a |
| 20. | Jasinski, M.; Jankowiak, A.; Januszko, A.; Bremer, M.; Pauluth, D.; Kaszynski, P. Liq. Cryst. 2008, 35, 343–350. doi:10.1080/02678290701817318 |
| 5. | Nagamine, T.; Januszko, A.; Ohta, K.; Kaszynski, P.; Endo, Y. Liq. Cryst. 2008, 35, 865–884. doi:10.1080/02678290802245450 |
| 40. | Kelker, H.; Scheurle, B. J. Phys. (Paris) 1969, 30-C4, 104–108. doi:10.1051/jphyscol:1969425 |
| 26. | Fox, M. A.; MacBride, J. A. H.; Peace, R. J.; Wade, K. J. Chem. Soc., Dalton Trans. 1998, 401–412. doi:10.1039/a707154j |
| 25. |
Kaszynski, P.; Kulikiewicz, K. K.; Januszko, A.; Douglass, A. G.; Tilford, R. W.; Pakhomov, S.; Patel, M. K.; Ke, Y.; Radziszewski, G. J.; Young, V. G., Jr.
submitted. |
| 44. | Onda, M.; Toda, A.; Mori, S.; Yamaguchi, I. J. Mol. Struct. 1986, 144, 47–51. doi:10.1016/0022-2860(86)80166-1 |
| 28. | Coult, R.; Fox, M. A.; Gill, W. R.; Herbertson, P. L.; McBride, J. A. H.; Wade, K. J. Organomet. Chem. 1993, 462, 19–29. doi:10.1016/0022-328X(93)83337-U |
| 45. | Scharfenberg, P.; Rozsondai, B.; Hargittai, I. Z. Naturforsch., A 1980, 35, 431–436. |
| 46. | Haase, W.; Athanassopoulou, M. A. Crystal Structures of LC Mesogens. In Structure and Bonding; Mingos, D. M. P., Ed.; Springer: Berlin, 1999; Vol. 94, pp 139–197. |
| 35. | Leblanc, J. P.; Tessier, M.; Judas, D.; Friedrich, C.; Noël, C.; Maréchal, E. Macromolecules 1993, 26, 4391–4399. doi:10.1021/ma00069a001 |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 51. | Effective diameter d for the four structural elements: A, d = 7.43 Å; B, d = 7.17 Å; C, d = 6.72 Å; D, d = 5.03 Å. For details see footnote 33 in reference 16. |
| 2. | Kaszynski, P.; Januszko, A.; Ohta, K.; Nagamine, T.; Potaczek, P.; Young, V. G., Jr.; Endo, Y. Liq. Cryst. 2008, 35, 1169–1190. doi:10.1080/02678290802409775 |
| 1. | Kaszynski, P.; Pakhomov, S.; Tesh, K. F.; Young, V. G., Jr. Inorg. Chem. 2001, 40, 6622–6631. doi:10.1021/ic010663x |
| 7. | Piecek, W.; Kaufman, J. M.; Kaszynski, P. Liq. Cryst. 2003, 30, 39–48. doi:10.1080/0267829021000039669 |
| 30. | Chaumeil, H.; Le Drian, C.; Defoin, A. Synthesis 2002, 757–760. doi:10.1055/s-2002-25773 |
| 31. | Sinclair, D. J.; Sherburn, M. S. J. Org. Chem. 2005, 70, 3730–3733. doi:10.1021/jo050105q |
| 32. | Tao, X.; Zhao, Y.; Shen, D. Synlett 2004, 359–361. doi:10.1055/s-2003-44992 |
| 50. | Vill, V. LiqCryst 4.6 database (LCI Publisher GmbH, Hamburg, Germany, www.lci-publisher.com) and references therein. |
| 33. | Garrett, P. M.; Smart, J. C.; Hawthorne, M. F. J. Am. Chem. Soc. 1969, 91, 4707–4709. doi:10.1021/ja01045a021 |
| 16. | Januszko, A.; Glab, K. L.; Kaszynski, P.; Patel, K.; Lewis, R. A.; Mehl, G. H.; Wand, M. D. J. Mater. Chem. 2006, 16, 3183–3192. doi:10.1039/b600068a |
| 25. |
Kaszynski, P.; Kulikiewicz, K. K.; Januszko, A.; Douglass, A. G.; Tilford, R. W.; Pakhomov, S.; Patel, M. K.; Ke, Y.; Radziszewski, G. J.; Young, V. G., Jr.
submitted. |
| 47. | Toyne, K. J. Liquid Crystal Behavior in Relation to Molecular Structure. In Thermotropic Liquid Crystals; Gray, G. W., Ed.; Wiley: New York, 1987; pp 28–63. |
| 48. | Demus, D. Chemical Structure and Mesogenic Properties. In Handbook of Liquid Crystals; Demus, D.; Goodby, J. W.; Gray, G. W.; Spiess, H.-W.; Vill, V., Eds.; Wiley-VCH: New York, 1998; Vol. 1, pp 133–187. |
| 29. | Miyaura, N.; Yanagi, Y.; Suzuki, A. Synth. Commun. 1981, 11, 513–519. doi:10.1080/00397918108063618 |
| 49. | Demus, D.; Hauser, A. Molecular structure and thermodynamic properties of nematic liquid crystals. In Selected Topics In Liquid Crystal Research; Koswig, H.-D., Ed.; Akademie-Verlag: Berlin, 1990; pp 19–44. |
© 2009 Jankowiak et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)