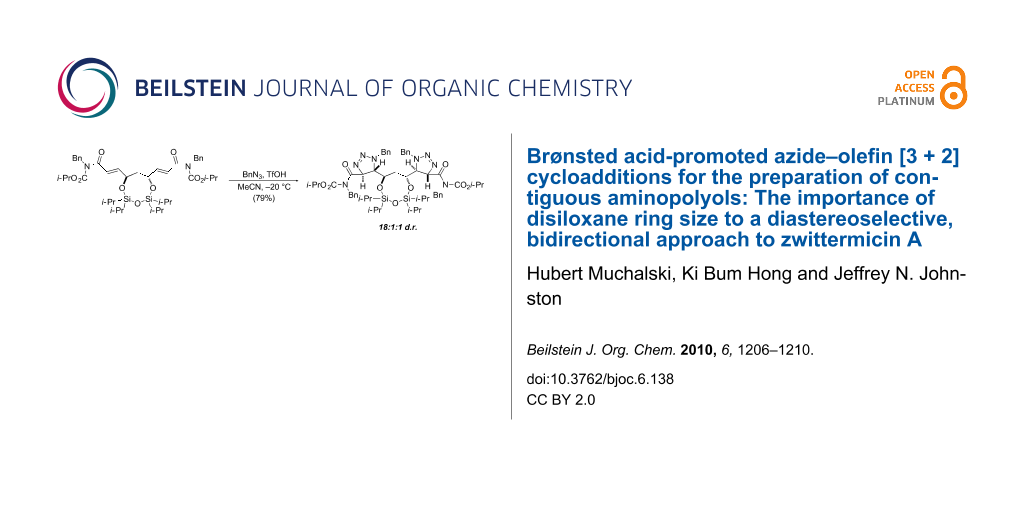
Abstract
We report the first study of substrate-controlled diastereoselection in a double [3 + 2] dipolar cycloaddition of benzyl azide with α,β-unsaturated imides. Using a strong Brønsted acid (triflic acid) to activate the electron deficient imide π-bond, high diastereoselection was observed provided that a 1,1,3,3-tetraisopropoxydisiloxanylidene group (TIPDS) is used to restrict the conformation of the central 1,3-anti diol. This development provides a basis for a stereocontrolled approach to the aminopolyol core of (−)-zwittermicin A using a bidirectional synthesis strategy.

Graphical Abstract
Introduction
Structural motifs such as 1,2-aminoalcohol, 1,2- and 1,3-diol are very prevalent features in natural products, especially polyketides. The structures of some of these, such as sorbistin A1 [1] or zwittermicin A [2], contain mostly aminopolyol moieties. Aminoalcohol and diol motifs are often constructed via alkene functionalization such as aminohydroxylation [3] and dihydroxylation [4] reactions, or by methods that forge the carbon–carbon bond such as the glycolate Mannich reaction [5]. Recently, we developed a Brønsted acid-promoted azide–olefin reaction as an alternative to metal catalyzed aminohydroxylations [6-8]. Triflic acid-promoted reaction of an alkyl azide with an α,β-unsaturated imide delivers a formal anti-aminohydroxylation product. We wondered whether azide–olefin functionalization could be used to prepare the complex aminopolyol core of zwittermicin A [9-12]. We were particularly intrigued by the possibility of a substrate controlled anti-diastereoselective azide–olefin reaction performed in a bidirectional fashion [13,14] to establish the requisite stereocenters of the C9–C15 C2 symmetric core of the natural product [11] as outlined in Scheme 1.
![[1860-5397-6-138-i1]](/bjoc/content/inline/1860-5397-6-138-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis outlining the stereocontrolled construction of the aminopolyol core of (−)-zwittermicin A using an azide–olefin double cycloaddition.
Scheme 1: Retrosynthetic analysis outlining the stereocontrolled construction of the aminopolyol core of (−)-...
Diastereoselective functionalization of the alkene of chiral allylic alcohols and ethers can be highly efficient, and the substrates are often easily accessible. The use of steric effects to achieve facial discrimination can be achieved by the introduction of a silyl group. Although several reports of diastereoselective azide–olefin cycloaddition reactions exist, they rely on intramolecular azide delivery under thermal conditions [15-21]. Examples of substrate control in an acid-catalyzed intermolecular reaction of azides with alkenes are limited [22]. We report our initial study of the intermolecular, diastereoselective sequence of two [3 + 2] cycloaddition reactions promoted by triflic acid where the large 1,3-diol protecting group – the 1,1,3,3-tetraisopropoxydisiloxanylidene group (TIPDS) – plays a crucial role in facial discrimination.
Results and Discussion
Our approach to the preparation of enantiomerically pure (−)-zwittermicin A (1) is based on a short synthesis of the C9–C15 aminopolyol core that takes advantage of its underlying C2 symmetry, as outlined in Scheme 1. Desymmetrization and functionalization of the bis(oxazolidine dione) 2 provides us with a foundation for the synthesis of 1 and would arise from the acid-promoted fragmentation of 2,3-anti / 2’,3’-anti bis(triazoline) 3 (Scheme 1). Compound 3 would be assembled in a sequential substrate-controlled intermolecular cycloaddition between imide 4 and benzyl azide.
We envisioned that the facial discrimination could be provided by a large alcohol protecting group at the central anti-1,3-diol (C11 and C13 in zwittermicin A). Our study of the diastereoselective reaction of benzyl azide with the bis(imide) is presented in Table 1. We began with a thermal reaction of the bis(imide) 5 that used the common di(tert-butylsilyl) functionality as a directing group [23-25]. After 45 min at 100 °C under microwave irradiation in neat benzyl azide [26], all three possible stereoisomers formed non-selectively (1:2:1 ratio). The desired 2,3-anti diastereomer 10a was separated and the relative stereochemistry was assigned through a spectroscopic study (NOESY, see Supporting Information File 1). Although the overall yield was satisfactory, purification of the desired 2,3-anti / 2’,3’-anti diastereomer 10a was tedious.
Table 1: Substrate-controlled double [3 + 2] cycloaddition.
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-138-i4.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Alkene | PG | R | Conditionsa | Product | a:b:cb | Yield (%)c |
| 1 | 5 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-138-i5.svg?max-width=637&scale=1.0)
|
Me | A | 10 | 1:2:1 | 72 |
| 2 | 5 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-138-i6.svg?max-width=637&scale=1.0)
|
Me | B | 10 | 1:2.5:5.4 | 67 |
| 3 | 6 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-138-i7.svg?max-width=637&scale=1.0)
|
i-Pr | B | 11 | 1:9:9 | 54 |
| 4 | 7 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-138-i8.svg?max-width=637&scale=1.0)
|
i-Pr | B | 12 | NDd | 7 |
| 5 | 8 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-6-138-i9.svg?max-width=637&scale=1.0)
|
Me | B | 13 | 18:1:1 | 27 |
| 6 | 9 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-6-138-i10.svg?max-width=637&scale=1.0)
|
i-Pr | B | 14 | 18:1:1 | 79 |
aConditions A: BnN3 (excess), microwave 100 °C, 1 h; Conditions B: TfOH (5 equiv), BnN3 (10 equiv) MeCN [0.2 M], −20 °C, 18 h. bRatio of products was measured using the 1H NMR of the crude reaction mixture. cCombined isolated yield. dND = not determined due to signal overlap in 1H NMR.
We then turned our attention to triflic acid-promoted triazoline formation. When imide 5 was reacted with BnN3 in the presence of triflic acid at −20 °C in acetonitrile, again, all three bis(triazolines) were isolated in 67% yield (Table 1, entry 2). In this experiment, however, the 2,3-syn diastereomer 10b was slightly favored and the desired 10a formed as a minor product. We found that a change of the ester group of the carbamate functionality from Me to i-Pr slightly improves selectivity. However, compound 6 led to mostly the undesired bis(triazolines) 11b and 11c (entry 3) in 54% combined yield whilst the desired product 11a was present in only trace amounts. Bis(imide) 7 with a smaller diisopropyl silyl ring decomposed under the reaction conditions and gave the mixture of triazolines in only 7% yield, the ratio of which could not be determined from the 1H NMR spectrum (Table 1, entry 4).
Our original expectation was that the siloxane protected 1,3-anti-diol would assume a twist-boat conformation in order to maintain its two alkene substituents in a pseudo-equatorial arrangement. We reasoned that expansion of the ring from six to eight members through the formation of a disiloxanylidene derivative might better achieve this goal by providing greater flexibility around the oxygen-substituted edge (Scheme 2).
![[1860-5397-6-138-i2]](/bjoc/content/inline/1860-5397-6-138-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Depictions of the likely major conformations of the siloxane (A) and disiloxane (B) rings.
Scheme 2: Depictions of the likely major conformations of the siloxane (A) and disiloxane (B) rings.
The 8-membered ring methyl carbamate 8 incorporating a tetraisopropoxydisiloxanylidene group [24,25] (TIPDS) was prepared. Not only did bis(imide) 8 provide the bis(triazoline) with high diastereoselection (Table 1, entry 5), it favored the desired anti,anti 13a (30% yield). Introduction of the isopropyl carbamate in bis(imide) 9 led to a significant increase in the yield of the 2,3-anti-bis(triazoline) 14 (79%) without loss of diastereoselection (Table 1, entry 6).
Due to the flexibility of the disiloxane ring we were unable to determine reliably the relative stereochemistry of 13 or 14 by NOE. However, bis(triazolines) 10a and 14a could be converted to the corresponding bis(oxazolidine diones) by treatment with triflic acid at room temperature (Scheme 3). The silyl protecting groups were removed with HF·pyridine in THF, and 15 and 16 converted to the same 1,3-diol 17 (Scheme 3).
![[1860-5397-6-138-i3]](/bjoc/content/inline/1860-5397-6-138-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Confirmation of the relative stereochemistry of bis(triazoline) 14a. (a) TfOH, CH3CN, rt, 18 h; (b) HF·pyridine, THF, 0 °C to rt, 1 h.
Scheme 3: Confirmation of the relative stereochemistry of bis(triazoline) 14a. (a) TfOH, CH3CN, rt, 18 h; (b)...
Conclusion
In summary, this first study of the substrate-controlled diastereoselective addition of benzyl azide to an unsaturated bis(imide) has demonstrated that high diastereoselection is possible using an anti-1,3-diol scaffold. However, it is important to protect this diol as an 8-membered dialkoxydisiloxane instead of a more traditional 6-membered dialkoxysilane. The anti,anti-selectivity observed in this transformation provides a foundation for the straightforward preparation of the aminopolyol backbone of (−)-zwittermicin A using a bidirectional chain functionalization strategy.
Supporting Information
| Supporting Information File 1: Experimental procedures, 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 620.2 KB | Download |
References
-
Ogawa, T.; Katano, K.; Matsui, M. Tetrahedron 1980, 36, 2727–2733. doi:10.1016/0040-4020(80)80148-7
Return to citation in text: [1] -
He, H.; Silo-Suh, L. A.; Handelsman, J.; Clardy, J. Tetrahedron Lett. 1994, 35, 2499–2502. doi:10.1016/S0040-4039(00)77154-1
Return to citation in text: [1] -
Bodkin, J. A.; McLeod, M. D. J. Chem. Soc., Perkin Trans. 1 2002, 2733–2746. doi:10.1039/B111276G
Return to citation in text: [1] -
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009
Return to citation in text: [1] -
Troyer, T. L.; Muchalski, H.; Johnston, J. N. Chem. Commun. 2009, 6195–6197. doi:10.1039/b913785h
Return to citation in text: [1] -
Mahoney, J. M.; Smith, C. R.; Johnston, J. N. J. Am. Chem. Soc. 2005, 127, 1354–1355. doi:10.1021/ja045608c
Return to citation in text: [1] -
Hong, K. B.; Donahue, M. G.; Johnston, J. N. J. Am. Chem. Soc. 2008, 130, 2323–2328. doi:10.1021/ja0779452
Return to citation in text: [1] -
Donahue, M. G.; Hong, K. B.; Johnston, J. N. Bioorg. Med. Chem. Lett. 2009, 19, 4971–4973. doi:10.1016/j.bmcl.2009.07.067
Return to citation in text: [1] -
Rogers, E. W.; Molinski, T. F. Org. Lett. 2007, 9, 437–440. doi:10.1021/ol062804a
Return to citation in text: [1] -
Rogers, E.; Dalisay, D.; Molinski, T. Angew. Chem., Int. Ed. 2008, 47, 8086–8089. doi:10.1002/anie.200801561
Return to citation in text: [1] -
Rogers, E. W.; Molinski, T. F. J. Org. Chem. 2009, 74, 7660–7664. doi:10.1021/jo901007v
Return to citation in text: [1] [2] -
Rogers, E. W.; Dalisay, D. S.; Molinski, T. F. Bioorg. Med. Chem. Lett. 2010, 20, 2183–2185. doi:10.1016/j.bmcl.2010.02.032
Return to citation in text: [1] -
Magnuson, S. Tetrahedron 1995, 51, 2167–2213. doi:10.1016/0040-4020(94)01070-G
Return to citation in text: [1] -
Poss, C. S.; Schreiber, S. L. Acc. Chem. Res. 1994, 27, 9–17. doi:10.1021/ar00037a002
Return to citation in text: [1] -
Norris, P.; Horton, D.; Giridhar, D. E. Tetrahedron Lett. 1996, 37, 3925–3928. doi:10.1016/0040-4039(96)00716-2
Return to citation in text: [1] -
Herdeis, C.; Schiffer, T. Synthesis 1997, 1405–1410. doi:10.1055/s-1997-1366
Return to citation in text: [1] -
Krülle, T. M.; de la Fuente, C.; Pickering, L.; Aplin, R. T.; Tsitsanou, K. E.; Zographos, S. E.; Oikonomakos, N. G.; Nash, R. J.; Griffiths, R. C.; Fleet, G. W. J. Tetrahedron: Asymmetry 1997, 8, 3807–3820. doi:10.1016/S0957-4166(97)00561-2
Return to citation in text: [1] -
Herdeis, C.; Schiffer, T. Tetrahedron 1999, 55, 1043–1056. doi:10.1016/S0040-4020(98)01085-0
Return to citation in text: [1] -
Broggini, G.; Garanti, L.; Molteni, G.; Pilati, T. Tetrahedron: Asymmetry 2001, 12, 1201–1206. doi:10.1016/S0957-4166(01)00190-2
Return to citation in text: [1] -
Broggini, G.; Marchi, I. D.; Martinelli, M.; Paladino, G.; Penoni, A. Lett. Org. Chem. 2004, 1, 221–223. doi:10.2174/1570178043400956
Return to citation in text: [1] -
Chen, M.; Gan, Y.; Harwood, L. M. Synlett 2008, 2008, 2119–2121. doi:10.1055/s-2008-1078595
Return to citation in text: [1] -
Reddy, D. S.; Judd, W. R.; Aubé, J. Org. Lett. 2003, 5, 3899–3902. doi:10.1021/ol0355130
Return to citation in text: [1] -
Corey, E. J.; Hopkins, P. B. Tetrahedron Lett. 1982, 23, 4871–4874. doi:10.1016/S0040-4039(00)85735-4
Return to citation in text: [1] -
Kocienski, P. Protecting groups; Georg Thieme: New York, 2004.
Return to citation in text: [1] [2] -
Wuts, P. Greene's protective groups in organic synthesis; Wiley-Interscience: Hoboken, N.Y., 2007.
Return to citation in text: [1] [2] -
Caution should always be exercised when azides are heated or treated with strong acid, but we have never observed an uncontrolled reaction or off-gas during our studies.
Return to citation in text: [1]
| 1. | Ogawa, T.; Katano, K.; Matsui, M. Tetrahedron 1980, 36, 2727–2733. doi:10.1016/0040-4020(80)80148-7 |
| 5. | Troyer, T. L.; Muchalski, H.; Johnston, J. N. Chem. Commun. 2009, 6195–6197. doi:10.1039/b913785h |
| 4. | Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009 |
| 3. | Bodkin, J. A.; McLeod, M. D. J. Chem. Soc., Perkin Trans. 1 2002, 2733–2746. doi:10.1039/B111276G |
| 26. | Caution should always be exercised when azides are heated or treated with strong acid, but we have never observed an uncontrolled reaction or off-gas during our studies. |
| 2. | He, H.; Silo-Suh, L. A.; Handelsman, J.; Clardy, J. Tetrahedron Lett. 1994, 35, 2499–2502. doi:10.1016/S0040-4039(00)77154-1 |
| 24. | Kocienski, P. Protecting groups; Georg Thieme: New York, 2004. |
| 25. | Wuts, P. Greene's protective groups in organic synthesis; Wiley-Interscience: Hoboken, N.Y., 2007. |
| 11. | Rogers, E. W.; Molinski, T. F. J. Org. Chem. 2009, 74, 7660–7664. doi:10.1021/jo901007v |
| 22. | Reddy, D. S.; Judd, W. R.; Aubé, J. Org. Lett. 2003, 5, 3899–3902. doi:10.1021/ol0355130 |
| 13. | Magnuson, S. Tetrahedron 1995, 51, 2167–2213. doi:10.1016/0040-4020(94)01070-G |
| 14. | Poss, C. S.; Schreiber, S. L. Acc. Chem. Res. 1994, 27, 9–17. doi:10.1021/ar00037a002 |
| 23. | Corey, E. J.; Hopkins, P. B. Tetrahedron Lett. 1982, 23, 4871–4874. doi:10.1016/S0040-4039(00)85735-4 |
| 24. | Kocienski, P. Protecting groups; Georg Thieme: New York, 2004. |
| 25. | Wuts, P. Greene's protective groups in organic synthesis; Wiley-Interscience: Hoboken, N.Y., 2007. |
| 9. | Rogers, E. W.; Molinski, T. F. Org. Lett. 2007, 9, 437–440. doi:10.1021/ol062804a |
| 10. | Rogers, E.; Dalisay, D.; Molinski, T. Angew. Chem., Int. Ed. 2008, 47, 8086–8089. doi:10.1002/anie.200801561 |
| 11. | Rogers, E. W.; Molinski, T. F. J. Org. Chem. 2009, 74, 7660–7664. doi:10.1021/jo901007v |
| 12. | Rogers, E. W.; Dalisay, D. S.; Molinski, T. F. Bioorg. Med. Chem. Lett. 2010, 20, 2183–2185. doi:10.1016/j.bmcl.2010.02.032 |
| 6. | Mahoney, J. M.; Smith, C. R.; Johnston, J. N. J. Am. Chem. Soc. 2005, 127, 1354–1355. doi:10.1021/ja045608c |
| 7. | Hong, K. B.; Donahue, M. G.; Johnston, J. N. J. Am. Chem. Soc. 2008, 130, 2323–2328. doi:10.1021/ja0779452 |
| 8. | Donahue, M. G.; Hong, K. B.; Johnston, J. N. Bioorg. Med. Chem. Lett. 2009, 19, 4971–4973. doi:10.1016/j.bmcl.2009.07.067 |
| 15. | Norris, P.; Horton, D.; Giridhar, D. E. Tetrahedron Lett. 1996, 37, 3925–3928. doi:10.1016/0040-4039(96)00716-2 |
| 16. | Herdeis, C.; Schiffer, T. Synthesis 1997, 1405–1410. doi:10.1055/s-1997-1366 |
| 17. | Krülle, T. M.; de la Fuente, C.; Pickering, L.; Aplin, R. T.; Tsitsanou, K. E.; Zographos, S. E.; Oikonomakos, N. G.; Nash, R. J.; Griffiths, R. C.; Fleet, G. W. J. Tetrahedron: Asymmetry 1997, 8, 3807–3820. doi:10.1016/S0957-4166(97)00561-2 |
| 18. | Herdeis, C.; Schiffer, T. Tetrahedron 1999, 55, 1043–1056. doi:10.1016/S0040-4020(98)01085-0 |
| 19. | Broggini, G.; Garanti, L.; Molteni, G.; Pilati, T. Tetrahedron: Asymmetry 2001, 12, 1201–1206. doi:10.1016/S0957-4166(01)00190-2 |
| 20. | Broggini, G.; Marchi, I. D.; Martinelli, M.; Paladino, G.; Penoni, A. Lett. Org. Chem. 2004, 1, 221–223. doi:10.2174/1570178043400956 |
| 21. | Chen, M.; Gan, Y.; Harwood, L. M. Synlett 2008, 2008, 2119–2121. doi:10.1055/s-2008-1078595 |
© 2010 Muchalski et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)