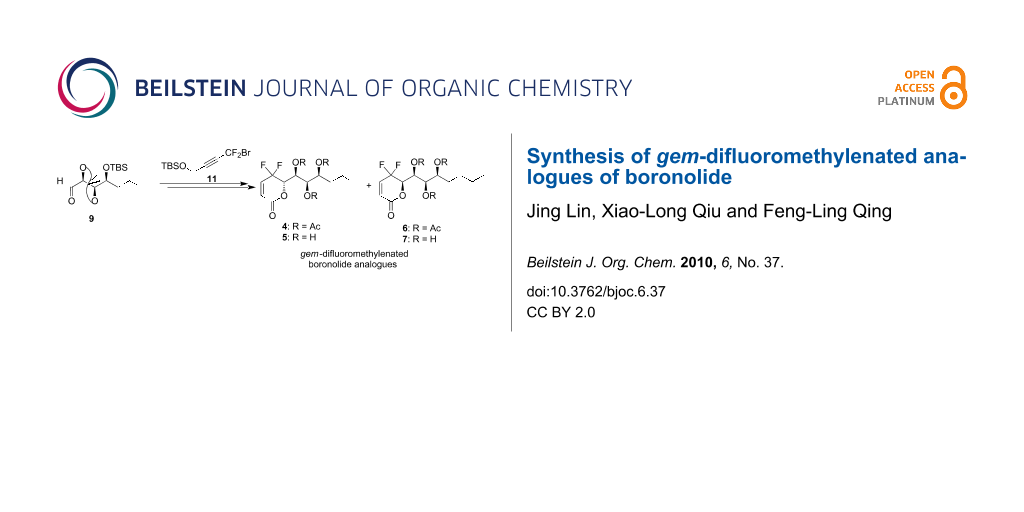
Abstract
The straightforward synthesis of four gem-difluoromethylenated analogues 4–7 of boronolide is described. The key steps of the synthesis include the concise preparation of the key intermediates 12a–b through the indium-mediated gem-difluoropropargylation of aldehyde 9 with the fluorine-containing building block 11 and the efficient construction of α,β-unsaturated-δ-lactones 15a–b via BAIB/TEMPO-procedure.

Graphical Abstract
Introduction
(+)-Boronolide (1), isolated from the bark and branches of Tetradenia fruticosa [1] and from the leaves of Tetradenia barberae [2], has been used as a traditional medicine in Madagascar and South Africa [2-4]. In addition, a partially deacetylated analogue 2 and the totally deacetylated analogue 3 have also been obtained from Tetradenia riparia [3,5], a Central African species traditionally employed by the Zulu as an emetic, and whose leaf infusions have also been reported to be effective against malaria [2,4]. Boronolide (1) and its analogues 2–3 feature an interesting polyacetoxylated (or polyhydroxyl) side chain and an α,β-unsaturated-δ-lactone moiety, making them an attractive target for total syntheses [6-16] since many natural products with a wide range of biological activity contain these structural elements. Noteworthily, structure–activity relationships have demonstrated that the α,β-unsaturated-δ-lactone moiety plays a key role in the bioactivity of many natural products. This is due to the fact that this unit is an excellent potential Michael acceptor for nucleophilic amino acid residues of the natural receptors interacting with these compounds [16-18]. Considering the similarity in size of fluorine and hydrogen atoms, the strong electron-withdrawing property of gem-difluoromethylene group (CF2) [19,20] and our continual efforts to prepare gem-difluoromethylenated analogues of natural products containing α,β-unsaturated-δ-lactone moiety [21-24], we intended to introduce a CF2 group to α,β-unsaturated-δ-lactone of boronolide at the γ-position (Figure 1). We envisioned that the resulting γ,γ-difluoromethylenylated–α,β-unsaturated conjugated double bond would be much more electron-deficient and therefore a better candidate to enhance the reactivity of the conjugated double bond as an acceptor with minimum steric change. In this article we describe the concise synthesis of gem-difluoromethylenated analogues 4–7 of boronolide.
![[1860-5397-6-37-1]](/bjoc/content/figures/1860-5397-6-37-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Boronolide (1), boronolide analogues 2–3 and gem-difluoromethylenated analogues 4–7.
Figure 1: Boronolide (1), boronolide analogues 2–3 and gem-difluoromethylenated analogues 4–7.
Results and Discussion
The retrosynthetic analysis of the target molecules 4–7 is outlined in Scheme 1. We envisioned that the key γ,γ-gem-difluoromethylenated α,β-unsaturated-δ-lactone scaffold could be constructed via an oxidation–cyclization reaction of intermediate A according to our published procedures [22]. cis-Selective reduction of homopropargyl alcohol B would provide the homoallylic alcohol A. The alcohol B could be obtained via an indium-mediated reaction of the fluorine-containing building block C and the protected aldehyde D, which in turn could be readily prepared by the reported procedure. Three chiral centres in our target molecules would be derived from D-glucono-δ-lactone, and the last one could be constructed by diastereoselective propargylation of the aldehyde.
![[1860-5397-6-37-i1]](/bjoc/content/inline/1860-5397-6-37-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of target molecules 4–7.
Scheme 1: Retrosynthetic analysis of target molecules 4–7.
According to the retrosynthetic analysis our synthesis embarked from aldehyde 9, which was prepared from commercially available D-glucono-δ-lactone (8) in six steps, based on the reported route [15] (Scheme 2). The synthesis of the fluorine-containing intermediate 11 was accomplished from propargyl alcohol (10) by our improved procedure [22]. With these two key fragments in hand, we focused our efforts on the gem-difluoropropargylation reaction. Utilizing Hammond’s reaction conditions [25], we were pleased to find that treatment of aldehyde 9 with compound 11 in the presence of indium with THF–H2O (1:4, v/v) as solvent smoothly gave the expected product 12a in 48% yield and 12b in 30% yield. More pleasing was the fact that the diastereomers 12a and 12b could be easily separated by silica gel chromatography. The assignment of the stereochemistry of the formed alcohol groups in 12a and 12b was based on the comparison of the 19F NMR spectra of compounds 5 and 6 with those of our synthesized gem-difluoromethylenated goniodiols. The absolute configuration of the formed alcohol was determined by X-ray crystallographic analysis [23]. Initial attempts to convert the triple bond in compound 12a into the cis double bond via hydrogenation in the presence of Lindlar catalyst were unsuccessful. Even with the addition of quinoline, these reactions only resulted in inseparable mixtures. Fortunately, hydrogenation proceeded well by means of Pd–BaSO4–quinoline system [26], leading to the expected alcohol 13a in 96% yield. Subsequent selective removal of the primary TBS group in 13a with D-camphor-10-sulfonic acid (CSA) yielded the diol 14a in 80% yield. As expected, treatment of compound 14a with 0.2 equiv of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 3.0 equiv of [bis(acetoxy)iodo]benzene (BAIB) in dichloromethane at room temperature afforded the desired α,β-unsaturated-δ-lactone 15a in 80% yield. Exposure of compound 15a to a solution of 6 M HCl in aqueous THF removed the protecting groups smoothly, and the deacetylated boronolide derivative 5 was obtained in 90% yield. Additionally, gem-difluoromethylenated boronolide 4 was prepared in good yield via treatment of compound 5 with Ac2O/DMAP/Et3N.
![[1860-5397-6-37-i2]](/bjoc/content/inline/1860-5397-6-37-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of target molecules 4–5.
Scheme 2: Synthesis of target molecules 4–5.
Using similar reaction conditions, gem-difluoromethylenated boronolide analogues6–7 were also synthesized from the intermediate 12b (Scheme 3).
![[1860-5397-6-37-i3]](/bjoc/content/inline/1860-5397-6-37-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of target molecules 6–7.
Scheme 3: Synthesis of target molecules 6–7.
Conclusion
In summary, we accomplished a concise synthesis of the gem-difluoromethylenated analogues of boronolide 4–7. Our approach featured the preparation of separable key diastereoisomers 12a–b from the indium-mediated gem-difluoropropargylation of aldehyde 9 with fluorine-containing building block 11 and the efficient construction of α,β-unsaturated-δ-lactones 15a–b via the BAIB/TEMPO-procedure.
Supporting Information
Supporting information features synthesis and characterization of gem-difluoromethylenated analogues of boronolide:
| Supporting Information File 1: Synthesis, characterization of concerned compounds. | ||
| Format: PDF | Size: 110.7 KB | Download |
References
-
Franca, N. C.; Polonsky, J. C. R. Seances Acad. Sci., Ser. C 1971, 273, 439–441.
Return to citation in text: [1] -
Davies-Coleman, M. T.; Rivett, D. E. A. Phytochemistry 1987, 26, 3047–3050. doi:10.1016/S0031-9422(00)84590-2
Return to citation in text: [1] [2] [3] -
Van Puyvelde, L.; De Kimpe, N.; Dube, S.; Chagnon-Dube, M.; Boily, Y.; Borremans, F.; Schamp, N.; Anteunis, M. J. O. Phytochemistry 1981, 20, 2753–2755. doi:10.1016/0031-9422(81)85280-6
Return to citation in text: [1] [2] -
Watt, J. M.; Brandwijk, M. G. B. The Medicinal and Poisonous Plants of Southern and Eastern Africa; Livingston: Edinburgh, 1962; p 516.
Return to citation in text: [1] [2] -
Van Puyvelde, L.; Dube, S.; Uwimana, E.; Uwera, C.; Domisse, R. A.; Esmans, E. L.; Van Schoor, O.; Vlietnick, A. Phytochemistry 1979, 18, 1215–1218. doi:10.1016/0031-9422(79)80138-7
Return to citation in text: [1] -
Jefford, C. W.; Moulin, M. C. Helv. Chim. Acta 1991, 74, 336–342. doi:10.1002/hlca.19910740213
Return to citation in text: [1] -
Nagano, H.; Yasui, H. Chem. Lett. 1992, 21, 1045–1048. doi:10.1246/cl.1992.1045
Return to citation in text: [1] -
Honda, T.; Horiuchi, S.; Mizutani, H.; Kanai, K. J. Org. Chem. 1996, 61, 4944–4948. doi:10.1021/jo960267+
Return to citation in text: [1] -
Chandrasekhar, M.; Raina, S.; Singh, V. K. Tetrahedron Lett. 2000, 41, 4969–4971. doi:10.1016/S0040-4039(00)00749-8
Return to citation in text: [1] -
Ghosh, A. K.; Bilcer, G. Tetrahedron Lett. 2000, 41, 1003–1006. doi:10.1016/S0040-4039(99)02246-7
Return to citation in text: [1] -
Murga, J.; Falomir, E.; Carda, M.; Marco, J. A. Tetrahedron: Asymmetry 2002, 13, 2317–2327. doi:10.1016/S0957-4166(02)00638-9
Return to citation in text: [1] -
Carda, M.; Rodriguez, S.; Segovia, B.; Marco, J. A. J. Org. Chem. 2002, 67, 6560–6563. doi:10.1021/jo025813f
Return to citation in text: [1] -
Trost, B. M.; Yeh, V. S. C. Org. Lett. 2002, 4, 3513–3516. doi:10.1021/ol026665i
Return to citation in text: [1] -
Chandrasekhar, M.; Chandra, K. L.; Singh, V. K. J. Org. Chem. 2003, 68, 4039–4045. doi:10.1021/jo0269058
Return to citation in text: [1] -
Hu, S.-G.; Hu, T.-S.; Wu, Y.-L. Org. Biomol. Chem. 2004, 2, 2305–2310. doi:10.1039/b406397j
Return to citation in text: [1] [2] -
Kumar, P.; Naidu, S. V. J. Org. Chem. 2006, 71, 3935–3941. doi:10.1021/jo0603781
Return to citation in text: [1] [2] -
de Fatima, A.; Kohn, L. K.; Antonio, M. A.; de Carvalho, J. E.; Pilli, R. A. Bioorg. Med. Chem. 2006, 14, 622–631. doi:10.1016/j.bmc.2005.08.036
Return to citation in text: [1] -
Buck, S. B.; Hardouin, C.; Ichikawa, S.; Soenen, D. R.; Gauss, C. M.; Hwang, I.; Swingle, M. R.; Bonness, K. M.; Honkanen, R. E.; Boger, D. L. J. Am. Chem. Soc. 2003, 125, 15694–15695. doi:10.1021/ja038672n
Return to citation in text: [1] -
Chambers, R. D. Fluorine in Organic Chemistry; Wiley: New York, 1973.
Return to citation in text: [1] -
Banks, R. E.; Smart, B. E.; Tatlow, J. C. Organofluorine Chemistry: Principles and Commercial Applications; Plenum Press: New York, 1994.
Return to citation in text: [1] -
You, Z.-W.; Wu, Y.-Y.; Qing, F.-L. Tetrahedron Lett. 2004, 45, 9479–9481. doi:10.1016/j.tetlet.2004.09.188
Return to citation in text: [1] -
You, Z.-W.; Jiang, Z.-X.; Wang, B.-L.; Qing, F.-L. J. Org. Chem. 2006, 71, 7261–7267. doi:10.1021/jo061012r
Return to citation in text: [1] [2] [3] -
You, Z.-W.; Zhang, X.; Qing, F.-L. Synthesis 2006, 2535–2542. doi:10.1055/s-2006-942467
Return to citation in text: [1] [2] -
Lin, J.; Yue, X.; Huang, P.; Cui, D.; Qing, F.-L. Synthesis 2010, 267–275. doi:10.1055/s-0029-1217099
Return to citation in text: [1] -
Wang, Z. G.; Hammond, G. B. J. Org. Chem. 2000, 65, 6547–6552. doi:10.1021/jo000832f
Return to citation in text: [1] -
Nakamura, Y.; Okada, M.; Sato, A.; Horikawa, H.; Koura, M.; Saito, A.; Taguchi, T. Tetrahedron 2005, 61, 5741–5753. doi:10.1016/j.tet.2005.04.034
Return to citation in text: [1]
| 1. | Franca, N. C.; Polonsky, J. C. R. Seances Acad. Sci., Ser. C 1971, 273, 439–441. |
| 2. | Davies-Coleman, M. T.; Rivett, D. E. A. Phytochemistry 1987, 26, 3047–3050. doi:10.1016/S0031-9422(00)84590-2 |
| 4. | Watt, J. M.; Brandwijk, M. G. B. The Medicinal and Poisonous Plants of Southern and Eastern Africa; Livingston: Edinburgh, 1962; p 516. |
| 26. | Nakamura, Y.; Okada, M.; Sato, A.; Horikawa, H.; Koura, M.; Saito, A.; Taguchi, T. Tetrahedron 2005, 61, 5741–5753. doi:10.1016/j.tet.2005.04.034 |
| 3. | Van Puyvelde, L.; De Kimpe, N.; Dube, S.; Chagnon-Dube, M.; Boily, Y.; Borremans, F.; Schamp, N.; Anteunis, M. J. O. Phytochemistry 1981, 20, 2753–2755. doi:10.1016/0031-9422(81)85280-6 |
| 5. | Van Puyvelde, L.; Dube, S.; Uwimana, E.; Uwera, C.; Domisse, R. A.; Esmans, E. L.; Van Schoor, O.; Vlietnick, A. Phytochemistry 1979, 18, 1215–1218. doi:10.1016/0031-9422(79)80138-7 |
| 2. | Davies-Coleman, M. T.; Rivett, D. E. A. Phytochemistry 1987, 26, 3047–3050. doi:10.1016/S0031-9422(00)84590-2 |
| 3. | Van Puyvelde, L.; De Kimpe, N.; Dube, S.; Chagnon-Dube, M.; Boily, Y.; Borremans, F.; Schamp, N.; Anteunis, M. J. O. Phytochemistry 1981, 20, 2753–2755. doi:10.1016/0031-9422(81)85280-6 |
| 4. | Watt, J. M.; Brandwijk, M. G. B. The Medicinal and Poisonous Plants of Southern and Eastern Africa; Livingston: Edinburgh, 1962; p 516. |
| 25. | Wang, Z. G.; Hammond, G. B. J. Org. Chem. 2000, 65, 6547–6552. doi:10.1021/jo000832f |
| 2. | Davies-Coleman, M. T.; Rivett, D. E. A. Phytochemistry 1987, 26, 3047–3050. doi:10.1016/S0031-9422(00)84590-2 |
| 23. | You, Z.-W.; Zhang, X.; Qing, F.-L. Synthesis 2006, 2535–2542. doi:10.1055/s-2006-942467 |
| 21. | You, Z.-W.; Wu, Y.-Y.; Qing, F.-L. Tetrahedron Lett. 2004, 45, 9479–9481. doi:10.1016/j.tetlet.2004.09.188 |
| 22. | You, Z.-W.; Jiang, Z.-X.; Wang, B.-L.; Qing, F.-L. J. Org. Chem. 2006, 71, 7261–7267. doi:10.1021/jo061012r |
| 23. | You, Z.-W.; Zhang, X.; Qing, F.-L. Synthesis 2006, 2535–2542. doi:10.1055/s-2006-942467 |
| 24. | Lin, J.; Yue, X.; Huang, P.; Cui, D.; Qing, F.-L. Synthesis 2010, 267–275. doi:10.1055/s-0029-1217099 |
| 15. | Hu, S.-G.; Hu, T.-S.; Wu, Y.-L. Org. Biomol. Chem. 2004, 2, 2305–2310. doi:10.1039/b406397j |
| 19. | Chambers, R. D. Fluorine in Organic Chemistry; Wiley: New York, 1973. |
| 20. | Banks, R. E.; Smart, B. E.; Tatlow, J. C. Organofluorine Chemistry: Principles and Commercial Applications; Plenum Press: New York, 1994. |
| 22. | You, Z.-W.; Jiang, Z.-X.; Wang, B.-L.; Qing, F.-L. J. Org. Chem. 2006, 71, 7261–7267. doi:10.1021/jo061012r |
| 16. | Kumar, P.; Naidu, S. V. J. Org. Chem. 2006, 71, 3935–3941. doi:10.1021/jo0603781 |
| 17. | de Fatima, A.; Kohn, L. K.; Antonio, M. A.; de Carvalho, J. E.; Pilli, R. A. Bioorg. Med. Chem. 2006, 14, 622–631. doi:10.1016/j.bmc.2005.08.036 |
| 18. | Buck, S. B.; Hardouin, C.; Ichikawa, S.; Soenen, D. R.; Gauss, C. M.; Hwang, I.; Swingle, M. R.; Bonness, K. M.; Honkanen, R. E.; Boger, D. L. J. Am. Chem. Soc. 2003, 125, 15694–15695. doi:10.1021/ja038672n |
| 6. | Jefford, C. W.; Moulin, M. C. Helv. Chim. Acta 1991, 74, 336–342. doi:10.1002/hlca.19910740213 |
| 7. | Nagano, H.; Yasui, H. Chem. Lett. 1992, 21, 1045–1048. doi:10.1246/cl.1992.1045 |
| 8. | Honda, T.; Horiuchi, S.; Mizutani, H.; Kanai, K. J. Org. Chem. 1996, 61, 4944–4948. doi:10.1021/jo960267+ |
| 9. | Chandrasekhar, M.; Raina, S.; Singh, V. K. Tetrahedron Lett. 2000, 41, 4969–4971. doi:10.1016/S0040-4039(00)00749-8 |
| 10. | Ghosh, A. K.; Bilcer, G. Tetrahedron Lett. 2000, 41, 1003–1006. doi:10.1016/S0040-4039(99)02246-7 |
| 11. | Murga, J.; Falomir, E.; Carda, M.; Marco, J. A. Tetrahedron: Asymmetry 2002, 13, 2317–2327. doi:10.1016/S0957-4166(02)00638-9 |
| 12. | Carda, M.; Rodriguez, S.; Segovia, B.; Marco, J. A. J. Org. Chem. 2002, 67, 6560–6563. doi:10.1021/jo025813f |
| 13. | Trost, B. M.; Yeh, V. S. C. Org. Lett. 2002, 4, 3513–3516. doi:10.1021/ol026665i |
| 14. | Chandrasekhar, M.; Chandra, K. L.; Singh, V. K. J. Org. Chem. 2003, 68, 4039–4045. doi:10.1021/jo0269058 |
| 15. | Hu, S.-G.; Hu, T.-S.; Wu, Y.-L. Org. Biomol. Chem. 2004, 2, 2305–2310. doi:10.1039/b406397j |
| 16. | Kumar, P.; Naidu, S. V. J. Org. Chem. 2006, 71, 3935–3941. doi:10.1021/jo0603781 |
| 22. | You, Z.-W.; Jiang, Z.-X.; Wang, B.-L.; Qing, F.-L. J. Org. Chem. 2006, 71, 7261–7267. doi:10.1021/jo061012r |
© 2010 Lin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)