Abstract
This article describes the chemical aminoacylation of the yeast phenylalanine suppressor tRNA with a series of amino acids bearing fluorinated side chains via the hybrid dinucleotide pdCpA and ligation to the corresponding truncated tRNA species. Aminoacyl-tRNAs can be used to synthesize biologically relevant proteins which contain fluorinated amino acids at specific sites by means of a cell-free translation system. Such engineered proteins are expected to contribute to our understanding of discrete fluorines’ interaction with canonical amino acids in a native protein environment and to enable the design of fluorinated proteins with arbitrary desired properties.
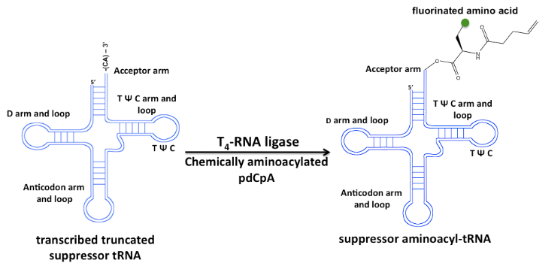
Graphical Abstract
Introduction
Over the past two decades, the interest in engineering proteins containing site-specific synthetic amino acids with novel functionalities has grown considerably. The utility of chemically aminoacylated suppressor transfer RNAs (tRNAs) combined with cell-free translation systems in producing proteins that contain non-canonical amino acids was reported independently from each other by Schultz and Chamberlin [1,2]. Their methodology is based on the following observations: 1) the central intermediate molecule in protein translation, the aminoacyl-tRNA (aa-tRNA) produced in the cell by specific tRNA synthetases (aaRSs) can be semi-synthesized; 2) a nonsense codon TAG can replace an amino acid-encoding codon at a desired position and can be recognized by the corresponding mutated orthogonal suppressor tRNA during the translation (Figure 1).
![[1860-5397-6-40-1]](/bjoc/content/figures/1860-5397-6-40-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: General strategy of using a chemically aminoacylated suppressor tRNA and an in vitro translation system to produce proteins which contain non-canonical amino acids (based on Noren et al. [1]). An amber stop codon, TAG, is introduced site-specifically into the protein-encoding gene, replacing an amino acid-encoding codon. The hybrid dinucleotide pdCpA is chemically aminoacylated with the non-canonical amino acid of interest and ligated to the appropriately truncated suppressor tRNA (tRNA-CA). Non-natural mutant proteins are produced by means of an in vitro protein synthesis system.
Figure 1: General strategy of using a chemically aminoacylated suppressor tRNA and an in vitro translation sy...
The key intermediates in this methodology are the suppressor aminoacyl-tRNAs. Due to the great number of reactive groups in the tRNA molecule, direct chemical acylation is not possible. Hecht and co-workers developed a procedure in which Nα-protected amino acids were used to chemically aminoacylate the dinucleotide pCpA. Subsequent enzymatic ligation to truncated tRNAs (without the 3′-terminal CA dinucleotide) yielded the desired AA-tRNAs [3]. More recently, this approach was optimized and the chemistry simplified by Schultz and co-workers. By using the hybrid dinucleotide pdCpA and activation of the amino acid as the cyanomethyl ester, selective coupling to the 2′- or 3′-hydroxyl group of the terminal adenosine was possible [4,5]. Since the Nα-aminoacyl moiety is not stable under the ligation conditions, the Nα-amino group of amino acid was either protected beforehand as the 6-nitroverastryloxycarbonyl (NVOC) derivative, a moiety which can be removed photochemically after the ligation, or left unprotected. However, in both cases low yields resulted. Lately, Hecht and co-workers reported the use of the N-(4-pentenoyl) protecting group as suitable for preparing a variety of misacylated tRNAs [6-9]. Removal of this group is achieved under mild chemical conditions by treatment with iodine solution; it has also been used in the preparation of caged proteins.
Fluorine is the most electronegative element and has a van der Waals radius of 1.47 Å [10]. Thus, substitution of a C–H bond with a C–F bond dramatically changes the electronic properties of the given molecule but exerts only a minor steric effect [11]. Due to the unique properties of the fluorine atom, the incorporation of amino acids which contain fluorinated side chains into peptides and proteins is becoming increasingly popular for the rational design of biopolymers and materials with novel biological properties. For example, certain fluorinated analogues of hydrophobic amino acids have been incorporated into the hydrophobic core of peptides, oligomers and proteins, leading to a significant increase in the thermal stability of the structure [12-15]. The introduction of fluoroalkyl groups into proteins can also enhance the hydrophobicity of the molecule, enabling better diffusion across the membranes [16]. Koksch and co-workers have developed a model peptide system based on the coiled-coil folding motif. They used it to show that the impact of fluorine substitution on structure and stability is strongly dependent on the position and the number of fluorine atoms within the peptide chain [17-19]. Finally, due to the high NMR sensitivity of fluorine, the incorporation of fluorinated amino acid analogues into proteins provides the opportunity for probing the structure and dynamics that play a role in protein–protein and protein–ligand interaction, and metabolic processes [20,21].
We report here the chemical and enzymatic aminoacylation of the yeast phenylalanine suppressor tRNA with a series of fluoroalkylated amino acids for site-specific protein mutagenesis (Figure 2). (RS)-2-amino-2-methyl-3,3,3-trifluoropropanoic acid (α-(Tfm)Ala) [22], (S)-ethylglycine (Abu) and two of its fluorinated analogues, (S)-2-amino-4,4-difluorobutanoic acid (DfeGly) [23] and (S)-2-amino-4,4,4-trifluorobutanoic acid (TfeGly) [24], were synthesized in the appropriate protected activated form and used to chemically aminoacylate tRNAPheCUA by means of the hybrid dinucleotide pdCpA and enzymatic ligation.
![[1860-5397-6-40-2]](/bjoc/content/figures/1860-5397-6-40-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of non-canonical amino acids. a. (S)-ethylglycine, b. (S)-2-amino-4,4-difluorobutanoic acid, c. (S)-2-amino-4,4,4-trifluorobutanoic acid, d. (RS)-2-amino-2-methyl-3,3,3-trifluoropropanoic acid.
Figure 2: Structures of non-canonical amino acids. a. (S)-ethylglycine, b. (S)-2-amino-4,4-difluorobutanoic a...
Results and Discussion
Syntheses of N-(4-pentenoyl) amino acid cyanomethyl esters
The aminoacylation of a suppressor tRNA is the first step to incorporate non-canonical amino acids into proteins. Several strategies have been developed to accomplish this [4-7,25-27]. We chose a combination of chemical and enzymatic aminoacylation which relies on the hybrid dinucleotide pdCpA and the T4-RNA ligase-mediated coupling to it to give a truncated suppressor tRNA. The Nα-amino groups of the amino acids were protected with the 4-pentenoyl group and the amino acids were activated as their corresponding cyanomethyl esters (Scheme 1).
![[1860-5397-6-40-i1]](/bjoc/content/inline/1860-5397-6-40-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General scheme for the synthesis of an N-(4-pentenoyl) amino acid cyanomethyl ester.
Scheme 1: General scheme for the synthesis of an N-(4-pentenoyl) amino acid cyanomethyl ester.
The N-(4-pentenoyl) protection of Abu and its fluorinated analogues DfeGly and TfeGly, and the preparation of their cyanomethyl esters were performed as described by Hecht and co-workers [7,9]. In the first step, the amino acid was treated with N-(4-pentenoyloxy)succinimide and in the second step treatment with iodoacetonitrile gave the desired compound in yields ranging from 59 to 81%. Due to the strong electron-withdrawing character of the C–F bond, the CF3 substituent in the α-position in α-(Tfm)Ala influences considerably the reactivity of both the amino and carboxylic groups; there is also a steric effect in this case. The amino group of α-(Tfm)Ala is generally protected by treatment with highly reactive mixed anhydrides or acid chlorides [28]. Thus, the N-(4-pentenoyl)-α-(Tfm)Ala was synthesized by means of 4-pentenoyl chloride. We investigated the reaction both in pyridine and DMF as the solvent, in the case of DMF, 4-dimethylaminopyridine (DMAP) was added as base. Although, pyridine also behaves as a base (pKa 5.21), higher yields were achieved with DMAP in DMF. The synthesis of N-(4-pentenoyl)-TfmAla cyanomethyl ester was achieved in an overall yield of 22%. The syntheses of N-(4-pentenoyl) amino acid cyanomethyl esters are summarized in Table 1.
Table 1: Syntheses of N-(4-pentenoyl) amino acid cyanomethyl esters.
| Yield of protection and activation (%) | 19F-NMR: δ (ppm)a | Mass (M+H)+ (m/z) (calculated) | |
|---|---|---|---|
| Abu (3a) | 81 | — | 225.1263 (224.1161) |
| DfeGly (3b) | 59 | −116.46 (tdd, 1F, J = 320.0 Hz, J = 58.6 Hz, J = 17.1 Hz), −115.70 (tdd, 1F, J = 320.0 Hz, J = 58.6 Hz, J = 17.1 Hz) | 261.1045 (260.0972) |
| TfeGly (3c) | 75 | −62.98 (t, 3F, J = 9.8 Hz) | 279.0929 (278.0878) |
| TfmAla (3d) | 22 | −76.227 (s, 3F) | 279.0929 (278.0878) |
as: singlet, t: triplet (See Supporting Information File 1).
Syntheses of 2′(3′)-O-[N-(4-pentenoyl)aminoacyl]-tRNAs and bis-2′,3′-O-[N-(4-pentenoyl)aminoacyl]-tRNAs
Chemical aminoacylation of pdCpA [4] was carried out using an N-protected amino acid activated as cyanomethyl ester in anhydrous DMF, and gave yields ranging from 40 to 90% (Scheme 2). The tetra-n-butylammonium (TBA) counter-ion is required to increase the solubility of pdCpA in DMF. Schultz and co-workers have reported that a ratio of 1:10 of TBA-pdCpA:activated ester results in the highly selective mono-acylation of the 2′,3′-hydroxyl groups of the adenosine ribose ring [5]. However, due to the expense of fluorinated amino acid analogues, we performed the aminoacylation reaction using a ratio of 1:2 or 1:3 TBA-pdCpA:activated ester at 40 °C overnight. Both mono-acylated and bis-acylated products were detected and purified by HPLC in the cases of Abu, TfeGly, and α-(Tfm)Ala, whereas only the mono-acylated product of DfeGly was observed. In general, longer incubation times and higher temperatures resulted in higher yields and increased amounts of bis-acylated products. A systematic investigation by Hecht and co-workers showed that such tandem activated tRNAs can also participate efficiently in the prokaryotic- and eukaryotic-based cell-free translation system. Both activated amino acids present in bis-acylated tRNAs can be recognized by the ribosome and incorporated into proteins [29,30]. pdCpA bearing Abu or the fluorinated amino acid analogue were efficiently ligated to the truncated suppressor tRNAPheCUA by treatment with T4-RNA ligase and were analyzed using denatured acidic PAGE (Figure 3) [31]. Thus, both our mono- and bis-acylated tRNAs are suitable for in vitro protein mutagenesis.
![[1860-5397-6-40-i2]](/bjoc/content/inline/1860-5397-6-40-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General scheme of synthesis of mono-2′(3′)-O-[N-(4-pentenoyl)aminoacyl]-pdCpAs and 2′-3′-bis-O-[N-(4-pentenoyl)aminoacyl]-pdCpAs.
Scheme 2: General scheme of synthesis of mono-2′(3′)-O-[N-(4-pentenoyl)aminoacyl]-pdCpAs and 2′-3′-bis-O-[N-(...
![[1860-5397-6-40-3]](/bjoc/content/figures/1860-5397-6-40-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Denaturing PAGE of ligation products of truncated suppressor tRNA and fluorinated aminoacyl-pdCpAs and Abu-pdCpA. Lane 1: RNA Marker 100 bp, Lane 2: transcribed full-length tRNACUA, Lane 3: transcribed truncated tRNACUA-COH, Lane 4: Abu-tRNACUA, Lane 5: DfeGly-tRNACUA, Lane 6: TfeGly-tRNACUA, Lane 7: α-(Tfm)Ala-tRNACUA. Amino groups are N-(4-pentenoyl) protected. Visualized by using Stains-all (Sigma-Aldrich®).
Figure 3: Denaturing PAGE of ligation products of truncated suppressor tRNA and fluorinated aminoacyl-pdCpAs ...
Conclusion and Outlook
The efficient chemical and enzymatic synthesis of three novel fluorinated aminoacyl-pdCpAs and Abu-pdCpA and their corresponding charged tRNAs is reported. These aminoacyl-tRNAs can be used for site-specific protein mutagenesis in a cell-free protein synthesis system and will enable a systematic investigation of the structural and dynamic behavior of fluorine within a native protein environment.
Supporting Information
Supporting information features detailed information on experimental procedures and compound characterization.
| Supporting Information File 1: Experimental procedures and compound characterization | ||
| Format: PDF | Size: 86.0 KB | Download |
Acknowledgements
We are grateful to Professor S. M. Hecht of the Department of Chemistry and Biology, Biodesign Institute, Arizona State University, AZ, USA for providing the tRNA gene. S. J. Y. thanks Dr. Cosimo D. Cadicamo for the preparation of DfeGly and very helpful discussions about chemical synthesis.
References
-
Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Science 1989, 244, 182–188. doi:10.1126/science.2649980
Return to citation in text: [1] [2] -
Bain, J. D.; Diala, E. S.; Glabe, C. G.; Dix, T. A.; Chamberlin, A. R. J. Am. Chem. Soc. 1989, 111, 8013–8014. doi:10.1021/ja00202a052
Return to citation in text: [1] -
Heckler, T. G.; Chang, L.-H.; Zama, Y.; Naka, T.; Chorgade, M. S.; Hecht, S. M. Biochemistry 1984, 23, 1468–1473. doi:10.1021/bi00302a020
Return to citation in text: [1] -
Robertson, S. A.; Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Nucleic Acids Res. 1989, 17, 9649–9660. doi:10.1093/nar/17.23.9649
Return to citation in text: [1] [2] [3] -
Robertson, S. A.; Ellman, J. A.; Schultz, P. G. J. Am. Chem. Soc. 1991, 113, 2722–2729. doi:10.1021/ja00007a055
Return to citation in text: [1] [2] [3] -
Lodder, M.; Golovine, S.; Hecht, S. M. J. Org. Chem. 1997, 62, 778–779. doi:10.1021/jo962170t
Return to citation in text: [1] [2] -
Lodder, M.; Wang, B. X.; Hecht, S. M. Methods 2005, 36, 245–251. doi:10.1016/j.ymeth.2005.04.002
Return to citation in text: [1] [2] [3] -
Choudhury, A. K.; Golovine, S. Y.; Dedkova, L. M.; Hecht, S. M. Biochemistry 2007, 46, 4066–4076. doi:10.1021/bi062042r
Return to citation in text: [1] -
Duca, M.; Chen, S. X.; Hecht, S. M. Methods 2008, 44, 87–99. doi:10.1016/j.ymeth.2007.10.007
Return to citation in text: [1] [2] -
Bondi, A. J. Phys. Chem. 1964, 68, 441–451. doi:10.1021/j100785a001
Return to citation in text: [1] -
Mikami, K.; Itoh, Y.; Yamanaka, M. Chem. Rev. 2004, 104, 1–16. doi:10.1021/cr030685w
Return to citation in text: [1] -
Tang, Y.; Ghirlanda, G.; Petka, W. A.; Nakajima, T.; DeGrado, W. F.; Tirrell, D. A. Angew. Chem., Int. Ed. 2001, 40, 1494–1496. doi:10.1002/1521-3773(20010417)40:8<1494::AID-ANIE1494>3.0.CO;2-X
Return to citation in text: [1] -
Bilgiçer, B.; Fichera, A.; Kumar, K. J. Am. Chem. Soc. 2001, 123, 4393–4399. doi:10.1021/ja002961j
Return to citation in text: [1] -
Chiu, H. P.; Suzuki, Y.; Gullickson, D.; Ahmad, R.; Kokona, B.; Fairman, R.; Cheng, R. P. J. Am. Chem. Soc. 2006, 128, 15556–15557. doi:10.1021/ja0640445
Return to citation in text: [1] -
Gottler, L. M.; Salud-Bea, R.; Marsh, E. N. Biochemistry 2008, 47, 4484–4490. doi:10.1021/bi702476f
Return to citation in text: [1] -
Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. ChemBioChem 2004, 5, 676–684. doi:10.1002/cbic.200400017
Return to citation in text: [1] -
Jäckel, C.; Koksch, B. Eur. J. Org. Chem. 2005, 21, 4483–4503. doi:10.1002/ejoc.200500205
Return to citation in text: [1] -
Jäckel, C.; Salwiczek, M.; Koksch, B. Angew. Chem., Int. Ed. 2006, 45, 4198–4203. doi:10.1002/anie.200504387
Return to citation in text: [1] -
Salwiczek, M.; Samsonov, S.; Vagt, T.; Nyakatura, E.; Fleige, E.; Numata, J.; Cölfen, H.; Pisabarro, M. T.; Koksch, B. Chemistry 2009, 15, 7628–7636. doi:10.1002/chem.200802136
Return to citation in text: [1] -
Hammill, J. T.; Miyake-Stoner, S.; Hazen, J. L.; Jackson, J. C.; Mehl, R. A. Nat. Protoc. 2007, 2, 2601–2607. doi:10.1038/nprot.2007.379
Return to citation in text: [1] -
Jackson, J. C.; Hammill, J. T.; Mehl, R. A. J. Am. Chem. Soc. 2007, 129, 1160–1166. doi:10.1021/ja064661t
Return to citation in text: [1] -
Smits, R.; Cadicamo, C. D.; Burger, K.; Koksch, B. Chem. Soc. Rev. 2008, 37, 1727–1739. doi:10.1039/b800310f
Return to citation in text: [1] -
Tsushima, T.; Kawada, K.; Ishihara, S.; Uchida, N.; Shiratori, O.; Higaki, J.; Hirata, M. Tetrahedron 1988, 44, 5375–5387. doi:10.1016/S0040-4020(01)86044-0
Return to citation in text: [1] -
Winkler, D.; Burger, K. Synthesis 1996, 1419–1421. doi:10.1055/s-1996-4409
Return to citation in text: [1] -
Hashimoto, N.; Ninomiya, K.; Endo, T.; Sisido, M. Chem. Commun. 2005, 4321–4323. doi:10.1039/b508194g
Return to citation in text: [1] -
Ninomiya, K.; Toshikazu, M.; Nishimura, M.; Sisido, M. J. Am. Chem. Soc. 2004, 126, 15984–15989. doi:10.1021/ja048200o
Return to citation in text: [1] -
Ohuchi, M.; Murakami, H.; Suga, T. Curr. Opin. Chem. Biol. 2007, 11, 537–542. doi:10.1016/j.cbpa.2007.08.011
Return to citation in text: [1] -
Sewald, N.; Hollweck, W.; Mütze, K.; Schierlinger, C.; Seymour, L. C.; Gaa, K.; Burger, K.; Koksch, B.; Jakubke, H. D. Amino Acids 1995, 8, 187–194. doi:10.1007/BF00806491
Return to citation in text: [1] -
Wang, B. X.; Zhou, J.; Lodder, M.; Anderson, R. D., III; Hecht, S. M. J. Biol. Chem. 2006, 281, 13865–13868. doi:10.1074/jbc.C600018200
Return to citation in text: [1] -
Duca, M.; Chen, S. X.; Hecht, S. M. Org. Biomol. Chem. 2008, 6, 3292–3299. doi:10.1039/b806790b
Return to citation in text: [1] -
Köhrer, C.; Rajbhandary, U. L. Methods 2008, 44, 129–138. doi:10.1016/j.ymeth.2007.10.006
Return to citation in text: [1]
| 5. | Robertson, S. A.; Ellman, J. A.; Schultz, P. G. J. Am. Chem. Soc. 1991, 113, 2722–2729. doi:10.1021/ja00007a055 |
| 28. | Sewald, N.; Hollweck, W.; Mütze, K.; Schierlinger, C.; Seymour, L. C.; Gaa, K.; Burger, K.; Koksch, B.; Jakubke, H. D. Amino Acids 1995, 8, 187–194. doi:10.1007/BF00806491 |
| 4. | Robertson, S. A.; Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Nucleic Acids Res. 1989, 17, 9649–9660. doi:10.1093/nar/17.23.9649 |
| 1. | Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Science 1989, 244, 182–188. doi:10.1126/science.2649980 |
| 2. | Bain, J. D.; Diala, E. S.; Glabe, C. G.; Dix, T. A.; Chamberlin, A. R. J. Am. Chem. Soc. 1989, 111, 8013–8014. doi:10.1021/ja00202a052 |
| 6. | Lodder, M.; Golovine, S.; Hecht, S. M. J. Org. Chem. 1997, 62, 778–779. doi:10.1021/jo962170t |
| 7. | Lodder, M.; Wang, B. X.; Hecht, S. M. Methods 2005, 36, 245–251. doi:10.1016/j.ymeth.2005.04.002 |
| 8. | Choudhury, A. K.; Golovine, S. Y.; Dedkova, L. M.; Hecht, S. M. Biochemistry 2007, 46, 4066–4076. doi:10.1021/bi062042r |
| 9. | Duca, M.; Chen, S. X.; Hecht, S. M. Methods 2008, 44, 87–99. doi:10.1016/j.ymeth.2007.10.007 |
| 4. | Robertson, S. A.; Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Nucleic Acids Res. 1989, 17, 9649–9660. doi:10.1093/nar/17.23.9649 |
| 5. | Robertson, S. A.; Ellman, J. A.; Schultz, P. G. J. Am. Chem. Soc. 1991, 113, 2722–2729. doi:10.1021/ja00007a055 |
| 6. | Lodder, M.; Golovine, S.; Hecht, S. M. J. Org. Chem. 1997, 62, 778–779. doi:10.1021/jo962170t |
| 7. | Lodder, M.; Wang, B. X.; Hecht, S. M. Methods 2005, 36, 245–251. doi:10.1016/j.ymeth.2005.04.002 |
| 25. | Hashimoto, N.; Ninomiya, K.; Endo, T.; Sisido, M. Chem. Commun. 2005, 4321–4323. doi:10.1039/b508194g |
| 26. | Ninomiya, K.; Toshikazu, M.; Nishimura, M.; Sisido, M. J. Am. Chem. Soc. 2004, 126, 15984–15989. doi:10.1021/ja048200o |
| 27. | Ohuchi, M.; Murakami, H.; Suga, T. Curr. Opin. Chem. Biol. 2007, 11, 537–542. doi:10.1016/j.cbpa.2007.08.011 |
| 4. | Robertson, S. A.; Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Nucleic Acids Res. 1989, 17, 9649–9660. doi:10.1093/nar/17.23.9649 |
| 5. | Robertson, S. A.; Ellman, J. A.; Schultz, P. G. J. Am. Chem. Soc. 1991, 113, 2722–2729. doi:10.1021/ja00007a055 |
| 7. | Lodder, M.; Wang, B. X.; Hecht, S. M. Methods 2005, 36, 245–251. doi:10.1016/j.ymeth.2005.04.002 |
| 9. | Duca, M.; Chen, S. X.; Hecht, S. M. Methods 2008, 44, 87–99. doi:10.1016/j.ymeth.2007.10.007 |
| 3. | Heckler, T. G.; Chang, L.-H.; Zama, Y.; Naka, T.; Chorgade, M. S.; Hecht, S. M. Biochemistry 1984, 23, 1468–1473. doi:10.1021/bi00302a020 |
| 23. | Tsushima, T.; Kawada, K.; Ishihara, S.; Uchida, N.; Shiratori, O.; Higaki, J.; Hirata, M. Tetrahedron 1988, 44, 5375–5387. doi:10.1016/S0040-4020(01)86044-0 |
| 1. | Noren, C. J.; Anthony-Cahill, S. J.; Griffith, M. C.; Schultz, P. G. Science 1989, 244, 182–188. doi:10.1126/science.2649980 |
| 16. | Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. ChemBioChem 2004, 5, 676–684. doi:10.1002/cbic.200400017 |
| 20. | Hammill, J. T.; Miyake-Stoner, S.; Hazen, J. L.; Jackson, J. C.; Mehl, R. A. Nat. Protoc. 2007, 2, 2601–2607. doi:10.1038/nprot.2007.379 |
| 21. | Jackson, J. C.; Hammill, J. T.; Mehl, R. A. J. Am. Chem. Soc. 2007, 129, 1160–1166. doi:10.1021/ja064661t |
| 12. | Tang, Y.; Ghirlanda, G.; Petka, W. A.; Nakajima, T.; DeGrado, W. F.; Tirrell, D. A. Angew. Chem., Int. Ed. 2001, 40, 1494–1496. doi:10.1002/1521-3773(20010417)40:8<1494::AID-ANIE1494>3.0.CO;2-X |
| 13. | Bilgiçer, B.; Fichera, A.; Kumar, K. J. Am. Chem. Soc. 2001, 123, 4393–4399. doi:10.1021/ja002961j |
| 14. | Chiu, H. P.; Suzuki, Y.; Gullickson, D.; Ahmad, R.; Kokona, B.; Fairman, R.; Cheng, R. P. J. Am. Chem. Soc. 2006, 128, 15556–15557. doi:10.1021/ja0640445 |
| 15. | Gottler, L. M.; Salud-Bea, R.; Marsh, E. N. Biochemistry 2008, 47, 4484–4490. doi:10.1021/bi702476f |
| 22. | Smits, R.; Cadicamo, C. D.; Burger, K.; Koksch, B. Chem. Soc. Rev. 2008, 37, 1727–1739. doi:10.1039/b800310f |
| 11. | Mikami, K.; Itoh, Y.; Yamanaka, M. Chem. Rev. 2004, 104, 1–16. doi:10.1021/cr030685w |
| 29. | Wang, B. X.; Zhou, J.; Lodder, M.; Anderson, R. D., III; Hecht, S. M. J. Biol. Chem. 2006, 281, 13865–13868. doi:10.1074/jbc.C600018200 |
| 30. | Duca, M.; Chen, S. X.; Hecht, S. M. Org. Biomol. Chem. 2008, 6, 3292–3299. doi:10.1039/b806790b |
| 17. | Jäckel, C.; Koksch, B. Eur. J. Org. Chem. 2005, 21, 4483–4503. doi:10.1002/ejoc.200500205 |
| 18. | Jäckel, C.; Salwiczek, M.; Koksch, B. Angew. Chem., Int. Ed. 2006, 45, 4198–4203. doi:10.1002/anie.200504387 |
| 19. | Salwiczek, M.; Samsonov, S.; Vagt, T.; Nyakatura, E.; Fleige, E.; Numata, J.; Cölfen, H.; Pisabarro, M. T.; Koksch, B. Chemistry 2009, 15, 7628–7636. doi:10.1002/chem.200802136 |
| 31. | Köhrer, C.; Rajbhandary, U. L. Methods 2008, 44, 129–138. doi:10.1016/j.ymeth.2007.10.006 |
© 2010 Ye et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)