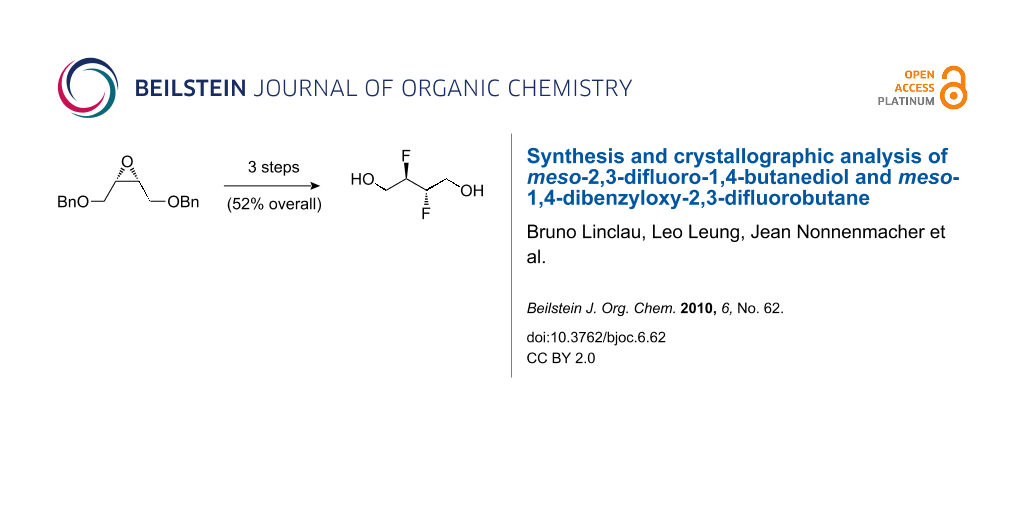
Abstract
A large-scale synthesis of meso-2,3-difluoro-1,4-butanediol in 5 steps from (Z)-but-2-enediol is described. Crystallographic analysis of the diol and the corresponding benzyl ether reveals an anti conformation of the vicinal difluoride moiety. Monosilylation of the diol is high-yielding but all attempts to achieve chain extension through addition of alkyl Grignard and acetylide nucleophiles failed.

Graphical Abstract
Introduction
Selective fluorination of bioactive compounds is a widely employed strategy for the modification of their properties [1]. Fluorine atoms can be introduced to modulate the pKa of adjacent acidic and basic functional groups as well as the lipophilicity, chemical and metabolic stability of the compound. Recent exciting reports describe weak but stabilising interactions between a C–F moiety and protein residues, which is certain to have implications in drug design [2,3]. Further important applications include molecular imaging using 18F [4], and modification of high-performance materials [5].
In recent years, the vicinal difluoride motif has received increasing attention due to the conformational properties instilled by the ‘gauche effect’ [6], which results in the vicinal difluoro gauche conformation being more stable than the corresponding anti conformation [7-9]. O’Hagan has demonstrated that vicinal difluoride substitution along a hydrocarbon chain of a fatty acid leads to conformational rigidity or disorder depending on the relative stereochemistry of the fluorine atoms, which originates from the enforcing or opposing fluorine gauche and hydrocarbon anti low-energy conformations [10]. As an extension, multi-vicinal tri- to hexafluorinated chains have been synthesised [11-16], which revealed yet another effect on the conformational behaviour, i.e. that conformations containing parallel 1,3-C–F bonds are destabilised. As an application, liquid crystals have been prepared containing a vicinal difluoride motif [14,17,18].
Efficient stereodefined synthesis of vicinal difluoride moieties is not straightforward. Direct methods include fluorination of alkenes with F2 [19], XeF2 [20], or hypervalent iodine species [21]. Such approaches often display poor stereoselectivity or result in rearrangement products. Treatment of 1,2-diols with SF4 [22,23], DAST [24], or deoxofluor [25] also leads to vicinal difluorides. Reaction with vicinal triflates has also been successful in some cases [7,26]. A common two-step method involves opening of an epoxide to give the corresponding fluorohydrin [27], followed by the conversion of the alcohol moiety to the fluoride [28]. Another two-step method is halofluorination of alkenes and subsequent halide substitution with silver fluoride [9,29,30].
The introduction of multiple fluorine atoms is often a cumbersome process, and in many cases a fluorinated building block approach [31,32] is more efficient. Known vicinal difluoride containing building blocks include (racemic) C2-symmetric and meso-2,3-difluorosuccinic acids (or esters) 1,2 (Figure 1) [9,22,23,33,34].
![[1860-5397-6-62-1]](/bjoc/content/figures/1860-5397-6-62-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Vicinal difluoride containing building blocks.
Figure 1: Vicinal difluoride containing building blocks.
Herein we describe the first synthesis of meso-2,3-difluoro-1,4-butanediol 3 as a further simple vicinal difluoride building block as well as its successful monosilylation, and our attempts to employ 3 for the synthesis of fluorinated hydrocarbons.
Results and Discussion
Synthesis
The synthesis of 3 was achieved from meso-epoxide 4, which was obtained from (Z)-2-butene-1,4-diol in excellent yield according to the published two-step sequence [35]. The optimisation of the reaction of 4 with fluoride sources is shown in Table 1.
Table 1: Conversion of epoxide 4 to the fluorohydrin.
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-62-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Reaction conditions | 5a | 6a | 4a |
|---|---|---|---|---|
| 1 | HF•py (70% HF), r.t., 3 h | 80b | – | – |
| 2 | KHF2, ethylene glycol, 150 °C, 3 h | 34 | 50 | – |
| 3 | KHF2, ethylene glycol, mol. Sieves, 150 °C, 3 h | – | c | – |
| 4 | KHF2, DMSO, 150 °C, 16 h | – | – | d |
| 5 | KHF2, DMF, 18-crown-6, reflux, 16 h | – | – | d |
| 6 | Bu4NH2F3 (1 equiv), xylene, reflux, 3 d | 11 | – | 57 |
| 7 | Bu4NH2F3 (1 equiv), KHF2 (1 equiv), 130 °C, 16 h | 71 | – | – |
| 8 | Bu4NH2F3 (1 equiv), KHF2 (1 equiv), 115 °C, 2.5 d | 91 | – | – |
a Isolated yield.
b Mixture of isomers.
c Complete conversion to 6 (TLC analysis).
d No reaction observed.
Reaction with Olah’s reagent [29] proceeded in excellent yield (Table 1, entry 1), however, the product was isolated as a mixture of isomers, which were not further characterised. Reaction with potassium hydrogen difluoride in ethylene glycol [36,37] gave the fluorohydrin in only modest yield (entry 2). Interestingly, the product arising from epoxide ring opening by ethylene glycol, 6, was isolated in 50% yield. The addition of molecular sieves (entry 3) led to complete conversion to 6 (TLC analysis). No reaction took place when DMSO (entry 4) or DMF/18-crown-6 were used as solvents [38,39] (entry 5). With Bu4NH2F3 as the fluoride source [40,41], 11% of the desired product (together with some elimination byproducts) was obtained when xylene was used as solvent (entry 6). However, reaction with a mixture of Bu4NH2F3 and KHF2 in the absence of solvent [42-44] led to an excellent 91% yield of the desired product 5 albeit after a relatively long reaction time (entry 8).
The subsequent conversion to 3 is shown in Scheme 1. Treatment of 5 with DAST in DCM at reflux temperature only gave 7 in 29% yield (not shown). A slight improvement (40% yield) was obtained when the reaction was conducted in hexane or toluene, but a procedure in which DAST was added to a solution of 5 in toluene at room temperature, followed by the addition of pyridine [28] and heating the reaction mixture for a prolonged period gave the desired vicinal difluoride in good yield. Nevertheless, while this procedure was deemed sufficiently safe to conduct at about the 50 mmol scale, further upscaling with a more thermally stable fluorinating reagent such as deoxofluor [45], Fluolead [46], or aminodifluorosulfinium tetrafluoroborate [47] would be recommended. Subsequent alcohol deprotection gave the target compound in almost quantitative yield in multigram quantities.
![[1860-5397-6-62-i1]](/bjoc/content/inline/1860-5397-6-62-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of meso-2,3-difluoro-1,4-butanediol.
Scheme 1: Synthesis of meso-2,3-difluoro-1,4-butanediol.
The potential of 3 as a building block, in particular for the construction of longer aliphatic chains of varying length, was investigated next. Thus (Scheme 2), the diol moiety in 3 was monoprotected as a silyl ether, and the remaining alcohol group was activated as the corresponding tosylate 9, triflate 10, mesylate 11, and bromide 12 as precursors for chain extension. Nucleophilic substitution of similar tosylates with phenolate nucleophiles has been previously described [18]. Reaction of 9–12 with a number of carbon nucleophiles was investigated.
![[1860-5397-6-62-i2]](/bjoc/content/inline/1860-5397-6-62-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Monoprotection of 3, and activation of the remaining alcohol.
Scheme 2: Monoprotection of 3, and activation of the remaining alcohol.
Unfortunately, reaction of 9–12 with alkyl Grignard and acetylide reagents did not lead to the desired chain extension. Reaction of 9 or 10 with a sodium or lithium acetylide led to decomposition, while 12 did not react under these conditions. Treatment of 11 with C9H19MgBr/CuBr was unsuccessful, whilst surprisingly, when 12 was subjected to this reagent combination (Scheme 3), the defluorinated reaction products 13 and 14 were obtained. We have not yet deduced an acceptable explanation for this unexpected result.
![[1860-5397-6-62-i3]](/bjoc/content/inline/1860-5397-6-62-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of 12 leading to defluorinated products.
Scheme 3: Reaction of 12 leading to defluorinated products.
Crystallographic analysis
Compounds 7 and 3 yielded colourless crystals suitable for study by single crystal X-ray diffraction [48]. The dibenzyl ether 7 crystallises in the monoclinic P21/c space group with half a molecule of 7 in the asymmetric unit. The molecule possesses crystallographic inversion symmetry. Two conformers are present in the crystal (55:45) which differ only in the sign of the torsion angle of the rings (Figure 2). The disparity in the amounts of each conformer present gives rise to the disorder observed in the crystal structure.
![[1860-5397-6-62-2]](/bjoc/content/figures/1860-5397-6-62-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular overlay of both conformers of 7.
Figure 2: Molecular overlay of both conformers of 7.
The vicinal difluoro group adopts an anti conformation with the F–C–C–F dihedral angle exactly 180°, which manifests itself in the crystallographic inversion centre. Nevertheless, each benzyloxy group does adopt a gauche conformation with its adjacent fluoro substituent where the F–C–C–O dihedral angle is 71.5°. Although strong H-bonding interactions are absent within the crystal, each molecule displays eight short contacts less than the sum of the van der Waals radii to its four nearest neighbours; three C–F···H–C contacts (2.554 Å, 2.581 Å and 2.637 Å) for each fluorine, and a pair of C–H···π contacts (2.662 Å to centroid of ring). The hydrogen atoms involved in the C–F contacts are an aromatic proton, the CHF and a CHHOBn proton (Figure 3).
![[1860-5397-6-62-3]](/bjoc/content/figures/1860-5397-6-62-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Crystal packing of 7 viewed along the b axis. Short contacts (see text) are shown in light blue.
Figure 3: Crystal packing of 7 viewed along the b axis. Short contacts (see text) are shown in light blue.
The diol 3 crystallises in the tetragonal space group I41/a with half a molecule of 3 in the asymmetric unit. This molecule also displays crystallographic inversion symmetry. In common with 7, the vicinal difluoro group of 3 adopts an anti conformation with a symmetry-constrained dihedral angle of 180°, and the hydroxyl groups adopt gauche conformations with the adjacent fluoro atoms with F–C–C–O dihedral angles of 66.8° (Figure 4).
![[1860-5397-6-62-4]](/bjoc/content/figures/1860-5397-6-62-4.png?scale=2.0&max-width=1024&background=FFFFFF)
There is strong hydrogen bonding between the hydroxyl groups of the molecule with each hydroxyl group acting both as donor and acceptor (O–H···O: 2.685 Å, 170.1°). The hydrogen bonded molecules are arranged helically about the crystallographic 41 screw axes. Thus the crystal structure comprises of alternating left and right handed hydrogen bonded helical constructs with each molecule part of two adjacent helices (Figure 5).
![[1860-5397-6-62-5]](/bjoc/content/figures/1860-5397-6-62-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Crystal packing of 3 viewed along the c axis. H-bonds are shown in light blue.
Figure 5: Crystal packing of 3 viewed along the c axis. H-bonds are shown in light blue.
Examination of the Cambridge Structural Database [49] (V5.31, November 2009) revealed three more meso-vic-difluoro compounds: 1,2-difluoro-1,2-diphenylethane, 2,3-difluorosuccinic acid and 2,3-difluorosuccinate benzylamide, all reported by O’Hagan [9]. Of these, only difluorosuccinic acid crystallises with the vicinal difluoro group in the expected gauche conformation, whilst both other structures, in common with the structures described in this work, contain the vicinal difluoro group in an anti conformation. The conformation of vicinal difluorides in solution can also be deduced from NMR studies. Schlosser has reported that the 3JH-F is around 22 Hz when the fluorines are in the syn configuration, because of a preferred gauche conformation, and around 14 Hz when in the anti configuration, because there is no overall preferred conformation [28]. Unfortunately, we were unable to extract 3JH-F values from the second order signals in both the 1H and 19F NMR spectra of 3 and 7, however, analysis of the coupling constants in 11 revealed two 3JH-F values of 10.1 and 9.6 Hz (3JF-F 13.5 Hz). Walba et al. have reported the 3JH-F values of a very similar syn-1-hydroxy-4-aryloxy-2,3-difluorobutane system to be around 22.0 Hz [17]. Hence, this value is indeed much higher than the 3JH-F values for 11, from which it can be concluded that the gauche effect in 11 (anti) is operating in solution.
Conclusion
The synthesis of meso-2,3-difluoro-1,4-butanediol 3 was achieved in 5 steps from (Z)-1,4-butenediol in 40% overall yield on a multigram scale. A high-yielding (94%) monosilylation was also achieved, but all attempts for chain extension met with failure. Crystallographic analysis revealed that the vicinal fluorine atoms in 3 and its dibenzyl ether 7 are in the anti conformation.
Experimental
1H and 13C NMR spectra were recorded at room temperature on a Bruker DPX400 or AV300 spectrometer as indicated. Low resolution ES mass and EIMS were recorded on a Waters ZMD and Thermoquest TraceMS quadrupole spectrometers, respectively. Infrared spectra were recorded as neat films on a Nicolet Impact 380 ATR spectrometer. Melting points were recorded on a Gallencamp Melting Point Apparatus and are uncorrected.
Column chromatography was performed on 230–400 mesh Matrex silica gel. Preparative HPLC was carried out using a Biorad Biosil D 90-10, 250 × 22 mm column eluting at 20 mL min−1, connected to a Kontron 475 refractive index detector. Reactions were monitored by TLC (Merck) with detection by KMnO4 or anisaldehyde stains.
Reaction solvents were dried before use as follows: THF and Et2O were distilled from sodium/benzophenone; CH2Cl2 and Et3N were distilled from CaH2; toluene was distilled from sodium.
X-ray data crystal structure analyses: Suitable crystals were selected and data collected on a Bruker Nonius Kappa CCD Area Detector equipped with a Bruker Nonius FR591 rotating anode (λ(MoKα) = 0.71073 Å) at 120 K driven by COLLECT [50] and processed by DENZO [51] software and corrected for absorption by using SADABS [52]. The structures were determined in SHELXS-97 and refined using SHELXL-97 [53]. All non-hydrogen atoms were refined anisotropically with hydrogen atoms included in idealised positions with thermal parameters riding on those of the parent atom.
syn-1,4-Bis(benzyloxy)-3-fluorobutan-2-ol (5)
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-62-i5.svg?max-width=637&scale=1.18182)
KHF2 (9.57g, 123 mmol) was added to a mixture of epoxide 4 (17.4 g, 61.3 mmol) and Bu4NH2F3 (10.6 g, 35.2 mmol) and the mixture stirred at 115 °C for 2.5 days. Et2O (300 mL) was added and the solution poured into sat. NaHCO3 (200 mL). The organic layer was washed successively with sat. NaHCO3 (100 mL) and brine (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography (EtOAc/petroleum ether 10% to 20%) to afford fluorohydrin 5 as a colourless oil (17.0 g, 91%). IR νmax (cm−1) 3062 w, 3030 w, 2993 w, 2858 w, 1496 w, 1453 m, 1369 w, 1088 s; 1H NMR (400 MHz, CDCl3) 7.42–7.20 (10H, m, ArH), 4.74 (1H, ddt, J = 47.5, 5.5, 3.5 Hz, CHF), 4.60 (1H, d, J = 12.0 Hz, CHaHbPh), 4.58 (1H, d, J = 12.0 Hz, CHcHdPh), 4.56 (1H, d, J = 12.0 Hz, CHaHbPh), 4.54 (1H, d, J = 12.0 Hz, CHcHdPh), 4.04 (1H, dm, J = 22.0 Hz, CHOH), 3.80 (1H, ddd, J = 23.0, 11.0, 4.0 Hz, CHaHbOBn), 3.76 (1H, ddd, J = 24.0, 11.0, 5.0 Hz, CHaHbOBn), 3.63 (1H, ddd, J = 10.0, 5.0, 1.0 Hz, CHcHdOBn), 3.59 (1H, ddd, J = 10.0, 6.5, 1.0 Hz, CHcHdOBn), 2.61 (1H, bd, J = 4.0 Hz, OH) ppm; 13C NMR (100 MHz, CDCl3) 137.9 (CAr), 137.7 (CAr), 128.6 (CHAr), 128.0 (CHAr), 127.9 (CHAr), 91.8 (d, J = 175.0 Hz, CHF), 73.9 (CH2Ph), 73.7 (CH2Ph), 70.37 (d, J = 5.5 Hz, CH2OBn), 70.34 (d, J = 20.0 Hz, CHOH), 69.8 (d, J = 23.0 Hz, CH2OBn) ppm; 19F NMR (282 MHz, CDCl3) −204.3 (1F, dq, J = 46.7, 23.4) ppm; ES+ m/z (%) 327 ((M+Na)+, 100); HRMS (ES+) for C18H21FO3Na (M+Na)+: Calcd 327.1367; Measured 327.1364.
Data for syn-3-(2-hydroxyethyl)-1,4-bis(benzyloxy)butan-2-ol (6)
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-62-i6.svg?max-width=637&scale=1.18182)
Colourless oil. IR νmax (cm−1) 3399 br, 3062 w, 3030 w, 2863 w, 1496 w, 1483 m, 1091 s; 1H NMR (400 MHz, CDCl3) 7.40–7.27 (10H, m), 4.54 (4H, s), 3.87 (1H, q, J = 5.5 Hz), 3.78–3.60 (7H, m), 3.58 (1H, dd, J = 10.0, 5.0 Hz), 3.51 (1H, dd, J = 9.5, 6.0 Hz), 3.25–2.30 (2H, br, OH) ppm; 13C NMR (100 MHz, CDCl3) 137.9 (CAr), 137.7 (CAr), 128.61 (CHAr), 128.59 (CHAr), 128.01 (CHAr), 127.99 (CHAr), 127.92 (CHAr), 79.4 (CHO), 73.7 (CH2Ph), 73.6 (CH2Ph), 73.2 (CH2O), 71.0 (CH2O), 70.9 (CHOH), 70.6 (CH2O), 62.3 (CH2O) ppm; ES+ m/z (%) 715 ((2M+Na)+, 20); HRMS (ES+) for C20H26O5Na (M+Na)+: Calcd 369.1672; Measured 369.1667.
meso-1,4-Bis(benzyloxy)-2,3-difluorobutane (7)
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-62-i7.svg?max-width=637&scale=1.18182)
DAST (9.6 mL, 72.7 mmol) was added to a solution of fluorohydrin 5 (17.0 g, 55.9 mmol) in toluene (75 mL) and the mixture stirred at r.t. for 5 min. Pyridine (11.9 mL, 145 mmol) was then added and the solution stirred at 70 °C for a further 16 h. The reaction mixture was cooled, poured into sat. NaHCO3 (100 mL) and Et2O (100 mL). The organic layer was washed successively with sat. NaHCO3 (100 mL) and brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude product was quickly purified by column chromatography (EtOAc/petroleum ether 0% to 5%) to afford a mixture which was recrystallised from hot petroleum ether. The filtrate was concentrated and recrystallised again from hot petroleum ether. The recrystallisation process was carried out for a third time to afford difluoride 7 as a white crystalline solid (overall yield 10.1 g, 59%). mp 56–57 °C; IR νmax (cm−1) 3058 w, 3030 w, 2916 w, 2878 w, 1607 w, 1496 w, 1449 m, 1137 s, 1048 s; 1H NMR (400 MHz, CDCl3) 7.40–7.27 (10H, m, ArH), 4.96–4.78 (2H, m, CHF × 2), 4.61 (4H, s, CH2Ph × 2), 3.88−3.71 (4H, m, CH2OBn) ppm; 13C NMR (100 MHz, CDCl3) 137.8 (CAr × 2), 128.6 (CHAr × 4), 128.0 (CHAr × 2), 127.8 (CHAr × 4), 90.0 (dd, J = 175.5, 27.5 Hz, ABX, 13CHF-12CHF × 2), 73.8 (CH2Ph × 2), 68.4 (m, ABX, 13CH2CHFCHF × 2) ppm; 19F NMR (282 MHz, CDCl3) −198.7 ppm; ES+ m/z (%) 329 ((M+Na)+, 100); HRMS (ES+) for C18H20F2O2Na (M+Na)+: Calcd 329.1324; Measured 329.1319.
meso-2,3-Difluorobutane-1,4-diol (3)
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-62-i8.svg?max-width=637&scale=1.18182)
Pd/C (5%; 13.9 g, 6.5 mmol) was added to a solution of difluoride 7 (10.0 g, 32.7 mmol) in THF (108 mL) and the mixture stirred at r.t. for 16 h under a H2 atmosphere (balloon). The suspension was filtered through celite, washed with MeOH and concentrated in vacuo. The crude product was purified by column chromatography (acetone/petroleum ether 30% to 50%) to afford diol 3 as a white crystalline solid (4.0 g, 97%). mp 99–101 °C; IR νmax (cm−1) 3329 br, 2936 br, 1647 br, 1042 s; 1H NMR (400 MHz, CDCl3) 4.85–4.70 (2H, m, CHF × 2), 4.08–3.83 (4H, m, CH2OH × 2), 1.92 (2H, t, J = 6.5 Hz, OH × 2) ppm; 13C NMR (100 MHz, acetone-d6) 92.6 (dd, J = 173.0, 26.0 Hz, ABX, 13CHF-12CHF × 2), 61.2 (m, ABX, 13CH2CHFCHF × 2) ppm; 19F{1H} NMR (282 MHz, acetone-d6) −200.5 ppm; HRMS (ES+) for C4H8F2O2Na (M+Na)+: Calcd 149.0385; Measured 149.0384.
anti-4-tert-Butyldimethylsilanyloxy-2,3-difluorobutan-1-ol (8)
![[Graphic 6]](/bjoc/content/inline/1860-5397-6-62-i9.svg?max-width=637&scale=1.18182)
NaH (60% dispersion in mineral oil; 1.40 g, 34.9 mmol) was added to a solution of diol 3 (4.0 g, 31.7 mmol) in THF (64 mL) and the mixture stirred at r.t. for 30 min. TBDMSCl (5.26 g, 34.9 mmol) was then added and the solution stirred at r.t. for a further 2 h. The reaction mixture was quenched with H2O (150 mL) and extracted with Et2O (200 mL × 3). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography (neat petroleum ether, then acetone/petroleum ether 10%) to afford silyl ether 8 as a colourless oil (7.14 g, 94%). IR νmax (cm−1) 3354 br, 2954 m, 2930 m, 2858 m, 1254 s, 1055 s; 1H NMR (400 MHz, CDCl3) 4.84–4.58 (2H, m, CHF × 2), 4.03–3.76 (4H, m, CH2O × 2), 2.47 (1H, br, OH), 0.91 (9H, s, SiC(CH3)3), 0.09 (6H, s, SiCH3 × 2) ppm; 1H{19F} NMR (400 MHz, CDCl3) 4.77 (1H, ddd, J = 6.0, 5.0, 3.0 Hz, CHF), 4.69 (1H, dt, J = 6.1, 3.5 Hz, CHF) ppm; 13C NMR (100 MHz, CDCl3) 90.8 (dd, J = 170.5, 21.0 Hz, CHF), 90.5 (dd, J = 178.5, 30.5 Hz, CHF), 61.7 (dd, J = 21.5, 5.0 Hz, CH2O), 61.3 (dd, J = 21.5, 5.0 Hz, CH2O), 25.9 (SiC(CH3)3), 18.4 (SiC), −5.38 (CH3), −5.43 (CH3) ppm; 19F NMR (376.5 MHz, CDCl3) −201.6 (d, J = 13.0 Hz), −201.9 (d, J = 13.0 Hz) ppm; ES+ m/z (%) 263 ((M+Na)+, 100); HRMS (ES+) for C10H22F2O2SiNa (M+Na)+: Calcd 263.1249; Measured 263.1256.
anti-4-tert-Butyldimethylsilanyloxy-2,3-difluorobutyl methanesulfonate (11)
![[Graphic 7]](/bjoc/content/inline/1860-5397-6-62-i10.svg?max-width=637&scale=1.18182)
MsCl (3.39 mL, 43.8 mmol) was added to a mixture of alcohol 8 (7.0 g, 29.2 mmol) and Et3N (6.6 mL, 46.7 mmol) in DCM (64 mL) and the mixture stirred at r.t. for 2 h. The reaction mixture was cooled to 0 °C, filtered, washed with cold Et2O/petroleum ether 1:1 and concentrated in vacuo. The crude product was purified by column chromatography (EtOAc/petroleum ether 15:85) to afford mesylate 11 as a colourless oil (9.29 g, 99%). [TLC monitoring should be performed using DCM/petroleum ether 6:4 until the complete consumption of the starting material, which has the same Rf value as the product when eluted with EtOAc/petroleum ether.] IR νmax (cm−1) 2955 m, 2931 m, 2858 m, 1473 w, 1360 s, 1256 m, 1178 s, 836 vs; 1H NMR (400 MHz, CDCl3) 4.98 (1H, ddtd, J = 46.9, 10.1, 6.6, 2.0 Hz, CHCH2OS), 4.68 (1H, dddt, J = 46.0, 9.6, 6.6, 3.3 Hz, CHCH2OSi), 4.62 (1H, ddt, J = 26.8, 12.1, 2.0 Hz, CHaHbOS), 4.49 (1H, dddd, J = 25.3, 12.1, 6.1, 2.0 Hz, CHaHbOS), 3.98 (1H, dddd, J = 18.5, 12.5, 3.5, 2.5 Hz, CHaHbOSi), 3.87 (1H, dddd, J = 30.5, 12.5, 3.5, 2.5 Hz, CHaHbOSi), 3.06 (3H, s, SCH3), 0.91 (9H, s, SiC(CH3)3), 0.09 (6H, s, SiCH3 × 2) ppm; 1H{19F} NMR (400 MHz, CDCl3) 4.98 (1H, td, J = 6.1, 2.0 Hz, CHCH2OS), 4.68 (1H, dt, J = 6.6, 3.0 Hz, CHCH2OSi) ppm; 13C NMR (100 MHz, CDCl3) 90.2 (dd, J = 176.5, 27.0 Hz, CHCH2OSi), 87.5 (dd, J = 177.0, 27.5 Hz, CHCH2OS), 67.8 (dd, J = 21.0, 6.0 Hz, CH2OS), 61.3 (dd, J = 21.5, 4.5 Hz, CH2OSi), 37.7 (SCH3), 25.9 (SiC(CH3)3), 18.4 (SiC), −5.4 (CH3), −5.5 (CH3) ppm; 19F{1H} NMR (282 MHz, CDCl3) −198.6 (d, 3JF-F = 13.5 Hz), −202.0 (d, 3JF-F = 13.5 Hz) ppm; ES+ m/z (%) 341 ((M+Na)+, 10); HRMS (ES+) for C11H24F2O4SSiNa (M+Na)+: Calcd 341.1025; Measured 341.1030.
anti-4-Bromo-2,3-difluoro-1-tert-butyldimethylsilanyloxybutane (12)
![[Graphic 8]](/bjoc/content/inline/1860-5397-6-62-i11.svg?max-width=637&scale=1.18182)
TBAB (9.94 g, 30.8 mmol) was added to a solution of mesylate 11 (8.91 g, 28.0 mmol) in THF (28 mL) and the mixture stirred at reflux for 3 h. The reaction mixture was concentrated in vacuo and the crude product purified by column chromatography (EtOAc/petroleum ether 0% to 25%) to afford bromide 12 as a yellow oil (6.95 g, 82%). IR νmax (cm−1) 2954 w, 2930 w, 2886 w, 2858 w, 1472 w, 1464 w, 1256 m, 836 vs, 778 s; 1H NMR (400 MHz, CDCl3) 4.90 (1H, ddtd, J = 46.0, 12.0, 6.5, 3.0 Hz, CHF), 4.66 (1H, dddt, J = 46.0, 9.0, 6.5, 3.5 Hz, CHF), 3.99 (1H, dddd, J = 19.5, 12.0, 3.0, 2.5 Hz, CHaHb), 3.89 (1H, dddd, J = 30.5, 12.5, 4.0, 3.0 Hz, CHaHb), 3.75 (1H, dddd, J = 23.5, 12.0, 3.0, 1.5 Hz, CHcHd), 3.63 (1H, dddd, J = 24.0, 12.0, 6.0, 2.0 Hz, CHcHd), 0.92 (9H, s, SiC(CH3)3), 0.10 (6H, s, SiCH3 × 2) ppm; 13C NMR (100 MHz, CDCl3) 91.0 (dd, J = 176.5, 27.0 Hz, CHF), 88.1 (dd, J = 177.0, 28.0 Hz, CHF), 61.3 (dd, J = 21.5, 4.0 Hz, CH2OSi), 30.4 (dd, J = 22.0, 4.5 Hz, CH2Br), 25.8 (SiC(CH3)3), 18.3 (SiC), −5.5 (CH3), −5.6 (CH3) ppm; 19F NMR (282 MHz, CDCl3) −192.6 (d, J = 15.0 Hz), −201.3 (d, J = 13.0 Hz) ppm; EI m/z (%) 245 ((M-tBu)+, 5), 303 and 305 (1:1, M+, 10).
(E)-1-tert-Butyldimethylsilanyloxytridec-2-ene (13) and (E)-1-bromo-4-tert-butyldimethylsilanyloxybut-2-ene (14)
![[Graphic 9]](/bjoc/content/inline/1860-5397-6-62-i12.svg?max-width=637&scale=1.18182)
C9H19MgBr (1.42 mL, 0.6M, solution in Et2O, 0.852 mmol) was added to a mixture of CuBr (137 mg, 0.955 mmol) in THF (1.2 mL). The mixture was then transferred to a solution of bromide 12 (140 mg, 0.462 mmol) in THF (1.2 mL) at 0 °C, warmed to r.t. and stirred for 3 h. The reaction mixture was quenched with H2O (10 mL) and extracted with Et2O (10 mL × 3). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography (DCM/petroleum ether 0% to 20%) to afford alkene 13 [54] as a mixture of isomers (1:11) as a yellow oil (24.1 mg, ~15%) and alkene 14 as a yellow oil (26.2 mg, ~20%) along with 72.0 mg (51%) of the starting bromide 12.
Alkene 13: IR νmax (cm−1) 2955 w, 2924 s, 2854 m, 1463 w, 1378 w, 834 s, 774 s; 1H NMR ((E)-isomer only, 400 MHz, CDCl3) 5.64 (1H, dtt, J = 15.5, 6.5, 1.5 Hz, CH=CH), 5.53 (1H, dtt, J = 15.0, 5.0, 1.0 Hz, CH=CH), 4.13 (2H, dq, J = 5.5, 1.5 Hz, CH2O), 2.06–2.00 (2H, m, CH2), 1.40–1.21 (16H, m, CH2 × 8), 0.92 (9H, s, SiC(CH3)3), 0.93–0.86 (3H, m, CH3), 0.08 (6H, s, SiCH3 × 2) ppm; 13C NMR (100 MHz, CDCl3) 131.8 (CH=CH), 129.3 (CH=CH), 64.3 (CH2O), 32.4 (CH2), 32.0 (CH2), 29.8 (CH2), 29.7 (CH2), 29.5 (CH2), 29.4 (CH2), 26.2 (SiC(CH3)3), 22.9 (CH2), 18.6 (SiC), 14.3 (CH3), −4.9 (SiCH3 × 2) ppm; EI m/z (%) 255.3 ((M-tBu)+, 57); HRMS (ES+) for C19H40OSiNa (M+Na)+: Calcd 335.2746; Measured 335.2741.
Alkene 14: Our spectra were in accord with literature copies of the spectra [55]: 1H NMR (400 MHz, CDCl3) 5.99−5.79 (2H, m, CH=CH), 4.21 (2H, ddd, J = 4.0, 2.5, 1.5 Hz, CH2), 3.98 (2H, ddd, J = 7.5, 2.0, 1.0 Hz, CH2), 0.92 (9H, s, SiC(CH3)3), 0.08 (6H, s, SiCH3 × 2) ppm; 13C NMR (100 MHz, CDCl3) 134.7 (CH=CH), 125.8 (CH=CH), 62.6 (CH2O), 32.4 (CH2Br), 25.9 (SiC(CH3)3), 18.4 (SiC), −5.3 (SiCH3 × 2); EI m/z (%) 207 and 209 ((M-tBu)+, 31, 1:1); HRMS (EI+) for C6H12O79BrSi (M-tBu)+: Calcd 206.9835; Measured 206.9841.
References
-
Ojima, I., Ed. Fluorine in Medicinal Chemistry and ChemicalBiology; Wiley-Blackwell: Chicester, U.K., 2009.
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Zürcher, M.; Diederich, F. J. Org. Chem. 2008, 73, 4345–4361. doi:10.1021/jo800527n
Return to citation in text: [1] -
Ametamey, S. M.; Honer, M.; Schubiger, P. A. Chem. Rev. 2008, 108, 1501–1516. doi:10.1021/cr0782426
Return to citation in text: [1] -
Babudri, F.; Farinola, G. M.; Naso, F.; Ragni, R. Chem. Commun. 2007, 1003–1022. doi:10.1039/b611336b
Return to citation in text: [1] -
Wolfe, S. Acc. Chem. Res. 1972, 5, 102–111. doi:10.1021/ar50051a003
Return to citation in text: [1] -
Angelini, G.; Gavuzzo, E.; Segre, A. L.; Speranza, M. J. Phys. Chem. 1990, 94, 8762–8766. doi:10.1021/j100388a004
Return to citation in text: [1] [2] -
Craig, N. C.; Chen, A.; Suh, K. H.; Klee, S.; Mellau, G. C.; Winnewisser, B. P.; Winnewisser, M. J. Am. Chem. Soc. 1997, 119, 4789–4790. doi:10.1021/ja963819e
Return to citation in text: [1] -
O’Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19
Return to citation in text: [1] [2] [3] [4] -
Tavasli, M.; O’Hagan, D.; Pearson, C.; Petty, M. C. Chem. Commun. 2002, 1226–1227. doi:10.1039/b202891c
Return to citation in text: [1] -
Nicoletti, M.; O’Hagan, D.; Slawin, A. M. Z. J. Am. Chem. Soc. 2005, 127, 482–483. doi:10.1021/ja045299q
Return to citation in text: [1] -
Hunter, L.; O’Hagan, D.; Slawin, A. M. Z. J. Am. Chem. Soc. 2006, 128, 16422–16423. doi:10.1021/ja066188p
Return to citation in text: [1] -
Hunter, L.; Slawin, A. M. Z.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2007, 46, 7887–7890. doi:10.1002/anie.200701988
Return to citation in text: [1] -
Nicoletti, M.; Bremer, M.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2007, 5075–5077. doi:10.1039/b711839b
Return to citation in text: [1] [2] -
Farran, D.; Slawin, A. M. Z.; Kirsch, P.; O’Hagan, D. J. Org. Chem. 2009, 74, 7168–7171. doi:10.1021/jo901360e
Return to citation in text: [1] -
Hunter, L.; Kirsch, P.; Slawin, A. M. Z.; O’Hagan, D. Angew. Chem., Int. Ed. 2009, 48, 5457–5460. doi:10.1002/anie.200901956
Return to citation in text: [1] -
Thurmes, W. M.; Wand, M. D.; Vohra, R. T.; Walba, D. M. Mol. Cryst. Liq. Cryst. 1991, 204, 1–7. doi:10.1080/00268949108046588
Return to citation in text: [1] [2] -
Vlahakis, J. Z.; Wand, M. D.; Lemieux, R. P. J. Am. Chem. Soc. 2003, 125, 6862–6863. doi:10.1021/ja0353309
Return to citation in text: [1] [2] -
Merritt, R. F. J. Am. Chem. Soc. 1967, 89, 609–612. doi:10.1021/ja00979a025
Return to citation in text: [1] -
Shieh, T.-C.; Yang, N. C.; Chernick, C. L. J. Am. Chem. Soc. 1964, 86, 5021–5022. doi:10.1021/ja01076a069
Return to citation in text: [1] -
Sawaguchi, M.; Hara, S.; Yoneda, N. J. Fluorine Chem. 2000, 105, 313–317. doi:10.1016/S0022-1139(99)00276-6
Return to citation in text: [1] -
Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7
Return to citation in text: [1] [2] -
Bell, H. M.; Hudlicky, M. J. Fluorine Chem. 1980, 15, 191–200. doi:10.1016/S0022-1139(00)82575-0
Return to citation in text: [1] [2] -
Meegalla, S. K.; Doller, D.; Liu, R.; Sha, D.; Lee, Y.; Soll, R. M.; Wisnewski, N.; Silver, G. M.; Dhanoa, D. Bioorg. Med. Chem. Lett. 2006, 16, 1702–1706. doi:10.1016/j.bmcl.2005.12.012
Return to citation in text: [1] -
Singh, R. P.; Shreeve, J. M. J. Fluorine Chem. 2002, 116, 23–26. doi:10.1016/S0022-1139(02)00065-9
Return to citation in text: [1] -
Marson, C. M.; Melling, R. C. Chem. Commun. 1998, 1223–1224. doi:10.1039/a801718b
Return to citation in text: [1] -
Haufe, G. J. Fluorine Chem. 2004, 125, 875–894. doi:10.1016/j.jfluchem.2004.01.023.
Review article.
Return to citation in text: [1] -
Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3
Return to citation in text: [1] [2] [3] -
Olah, G. A.; Welch, J. T.; Vankar, Y. D.; Nojima, M.; Kerekes, I.; Olah, J. A. J. Org. Chem. 1979, 44, 3872–3881. doi:10.1021/jo01336a027
Return to citation in text: [1] [2] -
Olah, G. A.; Nojima, M.; Kerekes, I. Synthesis 1973, 780–783. doi:10.1055/s-1973-22298
Return to citation in text: [1] -
Percy, J. M. Top. Curr. Chem. 1997, 193, 131–195. doi:10.1007/3-540-69197-9_4
Return to citation in text: [1] -
Paquette, L. A., Ed. Handbook of Reagents for Organic Synthesis. Fluorine-Containing Reagents; John Wiley & Sons: Chicester, U.K., 2007.
Return to citation in text: [1] -
Syvret, R. G.; Vassilaros, D. L.; Parees, D. M.; Pez, G. P. J. Fluorine Chem. 1994, 67, 277–282. doi:10.1016/0022-1139(93)02975-K
Return to citation in text: [1] -
Schüler, M.; O’Hagan, D.; Slawin, A. M. Z. Chem. Commun. 2005, 4324–4326. doi:10.1039/b506010a
Return to citation in text: [1] -
Garner, P.; Park, J. M. Synth. Commun. 1987, 17, 189–194. doi:10.1080/00397918708057220
Return to citation in text: [1] -
Umemura, E.; Tsuchija, T.; Kobayashi, Y.; Tanaka, K. Carbohydr. Res. 1992, 224, 141–163. doi:10.1016/0008-6215(92)84101-W
Return to citation in text: [1] -
Huang, J.-T.; Chen, L.-C.; Wang, L.; Kim, M.-H.; Warshaw, J. A.; Armstrong, D.; Zhu, Q.-Y.; Chou, T.-C.; Watanabe, K. A.; Matulic-Adamic, J.; Su, T.-L.; Fox, J. J.; Polsky, B.; Baron, P. A.; Gold, J. W. M.; Hardy, W. D.; Zuckerman, E. J. Med. Chem. 1991, 34, 1640–1646. doi:10.1021/jm00109a017
Return to citation in text: [1] -
Yang, S. S.; Min, J. M.; Beattie, T. R. Synth. Commun. 1988, 18, 899–905. doi:10.1080/00397918808060873
Return to citation in text: [1] -
Sattler, A.; Haufe, G. J. Fluorine Chem. 1994, 69, 185–190. doi:10.1016/0022-1139(94)03077-4
Return to citation in text: [1] -
Landini, D.; Molinari, H.; Penso, M.; Rampoldi, A. Synthesis 1988, 953–955. doi:10.1055/s-1988-27763
Return to citation in text: [1] -
Albert, P.; Cousseau, J. J. Chem. Soc., Chem. Commun. 1985, 961–962. doi:10.1039/C39850000961
Return to citation in text: [1] -
Landini, D.; Penso, M. Tetrahedron Lett. 1990, 31, 7209–7212. doi:10.1016/S0040-4039(00)97281-2
Return to citation in text: [1] -
Landini, D.; Albanese, D.; Penso, M. Tetrahedron 1992, 48, 4163–4170. doi:10.1016/S0040-4020(01)92194-5
Return to citation in text: [1] -
Lundt, I.; Albanese, D.; Landini, D.; Penso, M. Tetrahedron 1993, 49, 7295–7300. doi:10.1016/S0040-4020(01)87207-0
Return to citation in text: [1] -
Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048–7054. doi:10.1021/jo990566+
Return to citation in text: [1] -
Umemoto, T. 4-tert-Butyl-2,6-dimethylphenylsulfur trifluoride (FLUOLEAD™): a novel new fluorinating agent with high stability and ease of handling. Book of Abstracts, 19th International Symposium on Fluorine Chemistry; Jackson Hole: WY, U.S.A., 2009; pp 34 ff.
August 23–28.
Return to citation in text: [1] -
Beaulieu, F.; Beauregard, L.-P.; Courchesne, G.; Couturier, M.; LaFlamme, F.; L’Heureux, A. Org. Lett. 2009, 11, 5050–5053. doi:10.1021/ol902039q
Return to citation in text: [1] -
CCDC 767411 and CCDC 767412 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif
Return to citation in text: [1] -
Allen, F. H. Acta Crystallogr., Sect. B 2002, 58, 380–388. doi:10.1107/S0108768102003890
Return to citation in text: [1] -
Collect: Data collection software; The Netherlands, 1998.
Return to citation in text: [1] -
Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, Part A; Carter, C. W., Jr.; Sweet, R. M., Eds.; Methods in Enzymology, Vol. 276; Academic Press: NewYork, 1997; pp 307–326.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 1990, 46, 467–473. doi:10.1107/S0108767390000277
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] -
Ko, S. Y. Tetrahedron Lett. 1994, 35, 3601–3604. doi:10.1016/S0040-4039(00)73251-5
Return to citation in text: [1] -
DeBoef, B.; Counts, W. R.; Gilbertson, S. R. J. Org. Chem. 2007, 72, 799–804. doi:10.1021/jo0620462
Return to citation in text: [1]
| 28. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3 |
| 45. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048–7054. doi:10.1021/jo990566+ |
| 46. |
Umemoto, T. 4-tert-Butyl-2,6-dimethylphenylsulfur trifluoride (FLUOLEAD™): a novel new fluorinating agent with high stability and ease of handling. Book of Abstracts, 19th International Symposium on Fluorine Chemistry; Jackson Hole: WY, U.S.A., 2009; pp 34 ff.
August 23–28. |
| 1. | Ojima, I., Ed. Fluorine in Medicinal Chemistry and ChemicalBiology; Wiley-Blackwell: Chicester, U.K., 2009. |
| 25. | Singh, R. P.; Shreeve, J. M. J. Fluorine Chem. 2002, 116, 23–26. doi:10.1016/S0022-1139(02)00065-9 |
| 17. | Thurmes, W. M.; Wand, M. D.; Vohra, R. T.; Walba, D. M. Mol. Cryst. Liq. Cryst. 1991, 204, 1–7. doi:10.1080/00268949108046588 |
| 5. | Babudri, F.; Farinola, G. M.; Naso, F.; Ragni, R. Chem. Commun. 2007, 1003–1022. doi:10.1039/b611336b |
| 7. | Angelini, G.; Gavuzzo, E.; Segre, A. L.; Speranza, M. J. Phys. Chem. 1990, 94, 8762–8766. doi:10.1021/j100388a004 |
| 26. | Marson, C. M.; Melling, R. C. Chem. Commun. 1998, 1223–1224. doi:10.1039/a801718b |
| 4. | Ametamey, S. M.; Honer, M.; Schubiger, P. A. Chem. Rev. 2008, 108, 1501–1516. doi:10.1021/cr0782426 |
| 22. | Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7 |
| 23. | Bell, H. M.; Hudlicky, M. J. Fluorine Chem. 1980, 15, 191–200. doi:10.1016/S0022-1139(00)82575-0 |
| 9. | O’Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19 |
| 2. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 3. | Zürcher, M.; Diederich, F. J. Org. Chem. 2008, 73, 4345–4361. doi:10.1021/jo800527n |
| 24. | Meegalla, S. K.; Doller, D.; Liu, R.; Sha, D.; Lee, Y.; Soll, R. M.; Wisnewski, N.; Silver, G. M.; Dhanoa, D. Bioorg. Med. Chem. Lett. 2006, 16, 1702–1706. doi:10.1016/j.bmcl.2005.12.012 |
| 28. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3 |
| 14. | Nicoletti, M.; Bremer, M.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2007, 5075–5077. doi:10.1039/b711839b |
| 17. | Thurmes, W. M.; Wand, M. D.; Vohra, R. T.; Walba, D. M. Mol. Cryst. Liq. Cryst. 1991, 204, 1–7. doi:10.1080/00268949108046588 |
| 18. | Vlahakis, J. Z.; Wand, M. D.; Lemieux, R. P. J. Am. Chem. Soc. 2003, 125, 6862–6863. doi:10.1021/ja0353309 |
| 20. | Shieh, T.-C.; Yang, N. C.; Chernick, C. L. J. Am. Chem. Soc. 1964, 86, 5021–5022. doi:10.1021/ja01076a069 |
| 48. | CCDC 767411 and CCDC 767412 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif |
| 11. | Nicoletti, M.; O’Hagan, D.; Slawin, A. M. Z. J. Am. Chem. Soc. 2005, 127, 482–483. doi:10.1021/ja045299q |
| 12. | Hunter, L.; O’Hagan, D.; Slawin, A. M. Z. J. Am. Chem. Soc. 2006, 128, 16422–16423. doi:10.1021/ja066188p |
| 13. | Hunter, L.; Slawin, A. M. Z.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2007, 46, 7887–7890. doi:10.1002/anie.200701988 |
| 14. | Nicoletti, M.; Bremer, M.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2007, 5075–5077. doi:10.1039/b711839b |
| 15. | Farran, D.; Slawin, A. M. Z.; Kirsch, P.; O’Hagan, D. J. Org. Chem. 2009, 74, 7168–7171. doi:10.1021/jo901360e |
| 16. | Hunter, L.; Kirsch, P.; Slawin, A. M. Z.; O’Hagan, D. Angew. Chem., Int. Ed. 2009, 48, 5457–5460. doi:10.1002/anie.200901956 |
| 21. | Sawaguchi, M.; Hara, S.; Yoneda, N. J. Fluorine Chem. 2000, 105, 313–317. doi:10.1016/S0022-1139(99)00276-6 |
| 49. | Allen, F. H. Acta Crystallogr., Sect. B 2002, 58, 380–388. doi:10.1107/S0108768102003890 |
| 10. | Tavasli, M.; O’Hagan, D.; Pearson, C.; Petty, M. C. Chem. Commun. 2002, 1226–1227. doi:10.1039/b202891c |
| 47. | Beaulieu, F.; Beauregard, L.-P.; Courchesne, G.; Couturier, M.; LaFlamme, F.; L’Heureux, A. Org. Lett. 2009, 11, 5050–5053. doi:10.1021/ol902039q |
| 7. | Angelini, G.; Gavuzzo, E.; Segre, A. L.; Speranza, M. J. Phys. Chem. 1990, 94, 8762–8766. doi:10.1021/j100388a004 |
| 8. | Craig, N. C.; Chen, A.; Suh, K. H.; Klee, S.; Mellau, G. C.; Winnewisser, B. P.; Winnewisser, M. J. Am. Chem. Soc. 1997, 119, 4789–4790. doi:10.1021/ja963819e |
| 9. | O’Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19 |
| 18. | Vlahakis, J. Z.; Wand, M. D.; Lemieux, R. P. J. Am. Chem. Soc. 2003, 125, 6862–6863. doi:10.1021/ja0353309 |
| 9. | O’Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19 |
| 29. | Olah, G. A.; Welch, J. T.; Vankar, Y. D.; Nojima, M.; Kerekes, I.; Olah, J. A. J. Org. Chem. 1979, 44, 3872–3881. doi:10.1021/jo01336a027 |
| 30. | Olah, G. A.; Nojima, M.; Kerekes, I. Synthesis 1973, 780–783. doi:10.1055/s-1973-22298 |
| 27. |
Haufe, G. J. Fluorine Chem. 2004, 125, 875–894. doi:10.1016/j.jfluchem.2004.01.023.
Review article. |
| 51. | Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, Part A; Carter, C. W., Jr.; Sweet, R. M., Eds.; Methods in Enzymology, Vol. 276; Academic Press: NewYork, 1997; pp 307–326. |
| 28. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3 |
| 52. | Sheldrick, G. M. Acta Crystallogr., Sect. A 1990, 46, 467–473. doi:10.1107/S0108767390000277 |
| 53. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 40. | Landini, D.; Molinari, H.; Penso, M.; Rampoldi, A. Synthesis 1988, 953–955. doi:10.1055/s-1988-27763 |
| 41. | Albert, P.; Cousseau, J. J. Chem. Soc., Chem. Commun. 1985, 961–962. doi:10.1039/C39850000961 |
| 42. | Landini, D.; Penso, M. Tetrahedron Lett. 1990, 31, 7209–7212. doi:10.1016/S0040-4039(00)97281-2 |
| 43. | Landini, D.; Albanese, D.; Penso, M. Tetrahedron 1992, 48, 4163–4170. doi:10.1016/S0040-4020(01)92194-5 |
| 44. | Lundt, I.; Albanese, D.; Landini, D.; Penso, M. Tetrahedron 1993, 49, 7295–7300. doi:10.1016/S0040-4020(01)87207-0 |
| 36. | Umemura, E.; Tsuchija, T.; Kobayashi, Y.; Tanaka, K. Carbohydr. Res. 1992, 224, 141–163. doi:10.1016/0008-6215(92)84101-W |
| 37. | Huang, J.-T.; Chen, L.-C.; Wang, L.; Kim, M.-H.; Warshaw, J. A.; Armstrong, D.; Zhu, Q.-Y.; Chou, T.-C.; Watanabe, K. A.; Matulic-Adamic, J.; Su, T.-L.; Fox, J. J.; Polsky, B.; Baron, P. A.; Gold, J. W. M.; Hardy, W. D.; Zuckerman, E. J. Med. Chem. 1991, 34, 1640–1646. doi:10.1021/jm00109a017 |
| 38. | Yang, S. S.; Min, J. M.; Beattie, T. R. Synth. Commun. 1988, 18, 899–905. doi:10.1080/00397918808060873 |
| 39. | Sattler, A.; Haufe, G. J. Fluorine Chem. 1994, 69, 185–190. doi:10.1016/0022-1139(94)03077-4 |
| 35. | Garner, P.; Park, J. M. Synth. Commun. 1987, 17, 189–194. doi:10.1080/00397918708057220 |
| 29. | Olah, G. A.; Welch, J. T.; Vankar, Y. D.; Nojima, M.; Kerekes, I.; Olah, J. A. J. Org. Chem. 1979, 44, 3872–3881. doi:10.1021/jo01336a027 |
| 31. | Percy, J. M. Top. Curr. Chem. 1997, 193, 131–195. doi:10.1007/3-540-69197-9_4 |
| 32. | Paquette, L. A., Ed. Handbook of Reagents for Organic Synthesis. Fluorine-Containing Reagents; John Wiley & Sons: Chicester, U.K., 2007. |
| 54. | Ko, S. Y. Tetrahedron Lett. 1994, 35, 3601–3604. doi:10.1016/S0040-4039(00)73251-5 |
| 9. | O’Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19 |
| 22. | Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7 |
| 23. | Bell, H. M.; Hudlicky, M. J. Fluorine Chem. 1980, 15, 191–200. doi:10.1016/S0022-1139(00)82575-0 |
| 33. | Syvret, R. G.; Vassilaros, D. L.; Parees, D. M.; Pez, G. P. J. Fluorine Chem. 1994, 67, 277–282. doi:10.1016/0022-1139(93)02975-K |
| 34. | Schüler, M.; O’Hagan, D.; Slawin, A. M. Z. Chem. Commun. 2005, 4324–4326. doi:10.1039/b506010a |
| 55. | DeBoef, B.; Counts, W. R.; Gilbertson, S. R. J. Org. Chem. 2007, 72, 799–804. doi:10.1021/jo0620462 |
© 2010 Linclau et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)