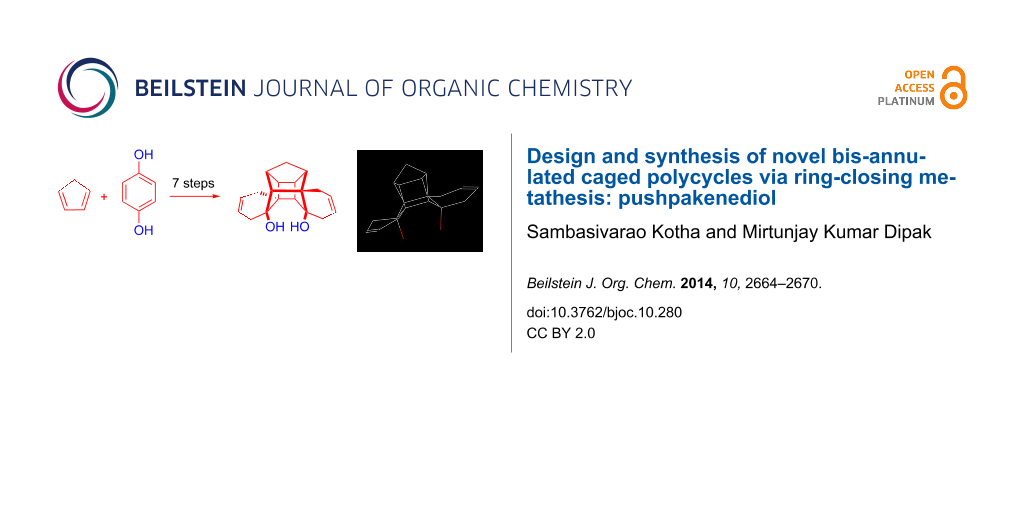
Abstract
Intricate caged molecular frameworks are assembled by an atom economical process via a Diels–Alder (DA) reaction, a Claisen rearrangement, a ring-closing metathesis (RCM) and an alkenyl Grignard addition. The introduction of olefinic moieties in the pentacycloundecane (PCUD) framework at appropriate positions followed by RCM led to the formation of novel heptacyclic cage systems.

Graphical Abstract
Introduction
Caged polycyclic compounds draw the attention of synthetic organic chemists due to their unusual reactivity patterns as well as their strained nature [1-9]. Several pentacycloundecane (PCUD) related molecules were found to be key structural elements in various drugs [10-12], high-energy materials [13-15] and supramolecules [16,17].
In addition, caged molecules possess unusual and often unique properties that are associated with their rigid carbocyclic framework [1,2]. They are useful synthons in the design and synthesis of natural as well as non-natural products [3,4]. Several intricate targets (Figure 1), such as snoutane (1) [18], basketane (2) [19], tetrahedrane (3) [20], triprismane [21], rocketene (4) [22], cubane (5) [23], garudane (6) [24], dodecahedrane (7) [25,26] and golcondane (8) [27] were assembled by employing various novel synthetic routes.
![[1860-5397-10-280-1]](/bjoc/content/figures/1860-5397-10-280-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected theoretically interesting molecules.
Figure 1: Selected theoretically interesting molecules.
Results and Discussion
In connection with our interest to prepare annulated PCUD, we proposed various dialkylated pentacyclic diones such as 1,9-dialkylpentacyclo[5.4.0.02,6.03,10.05,9]undeca-8,11-dione 12 by using [4 + 2] and [2 + 2] cycloaddition strategies, which involve the DA reaction of 2,5-dialkyl-1,4-benzoquinone 10 and 1,3-cyclopentadiene (9) followed by the formation of the cyclobutane ring through a [2 + 2] photocycloaddition reaction (Figure 2). Later on, one can introduce two allyl groups by using traditional carbonyl chemistry. The addition of alkyl groups can take place from the less hindered exo side of this caged system. If the R and R’ groups contain unsaturated systems one can think of constructing additional rings on the pentacyclic framework by utilizing ring-closing metathesis (RCM). By varying the length of unsaturated component one can generate diverse PCUD ring systems.
![[1860-5397-10-280-2]](/bjoc/content/figures/1860-5397-10-280-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic approach toward bis-annulated PCUD.
Figure 2: Retrosynthetic approach toward bis-annulated PCUD.
To realize the strategy depicted in Figure 2, we chose 2,5-diallyl-1,4-benzoquinone (18) as a viable option. We deliberately choose two unsaturated R groups at non-vicinal positions, because we have shown in an earlier report that the vicinal allyl groups in PCUD can be converted to a six-membered ring by RCM [28]. Since the two allyl groups in the PCUD framework are far apart, they did not undergo a RCM reaction. Later, by introducing additional unsaturated fragments in the PCUD system one can generate multiple rings in the PCUD system by using the RCM protocol. To this end, we began with the Claisen rearrangement of bis(allyloxy)benzene 15 to deliver the two possible rearranged diallylated products 16 and 17 [29,30] in equimolar ratio. When 2,5-diallyl-1,4-hydroquinone (17) was subjected to MnO2 oxidation in acetone at room temperature the corresponding 2,5-diallyl-1,4-benzoquinone (18) was obtained in good yield (67%). Since the quinone 18 is prone to polymerization (it gave a long streak on TLC when exposed to air at rt), it was immediately subjected to the [4 + 2] cycloaddition reaction with freshly prepared 1,3-cyclopentadiene (9) at 0–5 ºC to deliver compound 19 in 71.5% yield (Scheme 1).
![[1860-5397-10-280-i1]](/bjoc/content/inline/1860-5397-10-280-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthesis of diallylated tricyclic diene 19.
Scheme 1: The synthesis of diallylated tricyclic diene 19.
The formation of cycloadduct 19 was confirmed on the basis of 1H and 13C NMR spectral data. In 1H NMR data the integration of the olefinic region (ring olefinic proton) corresponds to the three protons and, as the cycloadduct is unsymmetrical, all the seventeen carbons appeared in the 13C NMR spectrum.
The stereochemistry of adduct 19 was expected to be endo as the reaction was performed at low temperature [28], and generally, the kinetically controlled product was produced under these reaction conditions. Our assumption was found to be correct, when we found that the DA adduct undergoes a smooth [2 + 2] photocyclization [31] upon exposure to UV light to generate caged dione 20 in good yield (80%). The structure of the photoadduct was assigned as 1,9-diallyl PCUD 20 on the basis of the spectral data (Scheme 2).
![[1860-5397-10-280-i2]](/bjoc/content/inline/1860-5397-10-280-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of diallylated pentacyclic dione 20.
Scheme 2: The synthesis of diallylated pentacyclic dione 20.
Having prepared the pentacyclic diallyldione 20, we ventured into the synthesis of PCUD based novel heptacyclic systems. The dione 20 was subjected to an allyl Grignard addition reaction, which resulted in the formation of tetra-allyldiol 21. The structure of diol 21 was established on the basis of high field 1H NMR (400 MHz) spectral data and further supported by 13C NMR spectral data (Scheme 3).
![[1860-5397-10-280-i3]](/bjoc/content/inline/1860-5397-10-280-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The synthesis of heptacyclic diol 22.
Scheme 3: The synthesis of heptacyclic diol 22.
The Grignard addition at a trigonal carbon may result in the formation of C–C bond in two possible ways (en face and zu face), but due to steric reasons only the exo–exo allyl addition product was isolated in the present case. The assumption of exo–exo stereochemistry has been further revived when the tetra-allyldiol 21 underwent a smooth RCM [32-37] upon its exposure to Grubbs’ second generation catalyst to deliver bis-annulated heptacyclic diol 22 in good yield (72%) (Scheme 3).
The molecular model and the optimized structure of heptacyclic diol 22 resembles the ancient flying machine “Puspak Viman” designed by the famous ancient aeronautical engineer saint Bhardwaj [38-40]. Therefore, we coined the name ‘pushpakenediol’ [41] for the heptacyclic diol. The optimization of the structure was carried out by means of Chem Draw 3D and the structure was visualized with the Mercury software (Figure 3).
![[1860-5397-10-280-3]](/bjoc/content/figures/1860-5397-10-280-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) Optimized structure of 22 (b) Ancient flying machine “Pushpak Viman”.
Figure 3: (a) Optimized structure of 22 (b) Ancient flying machine “Pushpak Viman”.
We turned our attention toward the next target, i.e., the symmetrical heptacyclic diol 27, by adopting the ring-rearrangement metathesis (RRM) protocol [42]. To this end, hexacyclic diones 23 [28] and 25 [28] were treated with an excess amount of allylmagnesium bromide (6 equiv) at room temperature to give the desired diallylated adducts 24 and 26 as sole products in 82% and 85% yield, respectively (Scheme 4). The structures of the adducts 24 and 26 were established based on high field 1H NMR (400 MHz) spectral data and further supported by 13C NMR spectral data.
![[1860-5397-10-280-i4]](/bjoc/content/inline/1860-5397-10-280-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The synthesis of diallylated hexacyclic diols.
Scheme 4: The synthesis of diallylated hexacyclic diols.
It was anticipated that the two allyl groups and the cyclohexene moiety present in 24 would undergo a ring-rearrangement metathesis (RRM), [42] which involves the ring-opening and ring-closing metathesis sequence (ROM–RCM) in a single step to generate the novel heptacyclic system 27. However, even under harsh reaction conditions, we did not observe the formation of the required RRM product (Scheme 5).
![[1860-5397-10-280-i5]](/bjoc/content/inline/1860-5397-10-280-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The attempted synthesis of heptacyclic diol via ring-rearrangement metathesis.
Scheme 5: The attempted synthesis of heptacyclic diol via ring-rearrangement metathesis.
The RRM reaction was carried out under various reaction conditions with different metathesis catalysts, for example, with Grubbs’ 1st and 2nd generation catalysts at rt as well as under reflux conditions. However, even the presence of ethylene during the metathesis sequence did not deliver the expected product, and the starting material remained unaltered (Scheme 5).
Conclusion
We demonstrated that the Grignard reaction in combination with the RCM reaction provides a useful strategy for the synthesis of novel and intricate molecular frameworks such as 22, which is suitable for studying stereoelectronic effects [43]. The strategy shown here is an atom economical process. The synthetic sequence opens up a new route to complex caged systems. At the same time non-participation of 24 in the RRM reaction reveals that systems which contain a sterically hindered cyclohexene ring are not suitable candidates for the tandem metathesis sequence.
Experimental
General
Reactions involving organometallic species were carried out under nitrogen by using oven-dried glassware and syringes. THF and Et2O were distilled from sodium/benzophenone under nitrogen immediately prior to use. Dichloromethane was distilled over P2O5. TLC was performed by using (10 × 5 cm) glass plates coated with Acme’s silica gel GF254 (containing 13% calcium sulfate as a binder). Flash-column chromatography was performed by using Aceme silica gel (100–200 mesh). Solvents were concentrated at reduced pressure on a Büchi R-114 rotary evaporator. 1H NMR (400 MHz) and 13C NMR (75.1 MHz) spectra were recorded at rt on a Bruker AX 400 with TMS (δ = 0.0 ppm, 1H NMR spectra) and CDCl3 (δ = 77.0 ppm, 13C NMR spectra) as internal standards. IR spectra were recorded on a Nicolet Impact-400 FTIR spectrometer. HRMS were determined on a Micromass Q-ToF spectrometer.
Materials
Grubbs’ 1st and 2nd generation and Grubbs–Hoveyda catalysts were purchased from Aldrich, Milwaukee (USA). Compounds 18, 19, 20, and 21 were prepared by procedures similar to those described in [28] for analogous compounds.
Preparation of 2,5-diallyl-1,4-benzoquinone (18)
To a solution of 2,5-diallyl-1,4-hydroquinone (17, 1.8 g, 9.45 mmol) in acetone (50 mL) was added MnO2 in excess (8 equiv) at rt. After completion of the reaction (TLC monitoring, 15 h), the reaction mixture was filtered off by using a pad of celite. The filtrate was evaporated to dryness and the residue was purified by distillation under reduced pressure at 106–107 °C (1 mmHg) to give 18 as a dark yellow oil (1.2 g, 67%). Bp: 106–107 °C at 1.0 mmHg (Lit. Bp: 105 °C at 1.0 mmHg) [16].
Preparation of 2,5-diallyltricyclo[6.2.1.02,7]undeca-4,9-diene-3,6-dione (19)
To a cooled solution (at 0 °C) of 2,5-diallyl-1,4-benzoquinone (18, 1.17 g, 6.21 mmol) in methanol (25 mL) was added freshly prepared 1,3-cyclopentadiene (0.48 mL, 6.24 mmol) in a dropwise manner. After completion of the reaction (TLC monitoring, 11 h), the solvent was evaporated under reduced pressure, and the residue was purified by silica-gel column chromatography (3% ethyl acetate/petroleum ether) to give 19 (1.13 g, 71.5%) as a thick yellow liquid. IR (Neat) νmax: 1337, 1666, 2991 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.4 (d, J = 8.8 Hz, 1H), 1.6 (d, J = 9.2 Hz, 1H), 2.16 (dd, J1 = 8.8 Hz, J2 = 8.2 Hz, 1H), 2.82 (dd, J1 = J2 = 6.7 Hz, 1H), 2.90–3.10 (m, 4H), 3.3 (s, 1H), 4.42–5.12 (m, 4H), 5.92–5.94 (m, 1H), 5.63–5.73 (m, 1H), 5.93 (dd, J1 = 2.0 Hz, J2 = 3.0 Hz, 1H), 6.05 (dd, J1 = 2.0 Hz, J2 = 3.0 Hz, 1H), 6.3 (s, 1H) ppm; 13C NMR (100.6 MHz, CDCl3) δ 32.8, 44.9, 46.4, 49.3, 53.1, 54.4, 57.4, 118.2, 132.8, 133.1, 134.9, 137.4, 139.55, 139.58, 152.3, 198.8, 202.0 ppm; HRMS (Q-ToF ES+) m/z: calcd for C17H18O2K, 293.0944; found, 293.1086 [M + K]+.
Preparation of 1,9-diallylpentacyclo[5.4.0.02,6.03,10.05,9]undeca-8,11-dione (20)
Tricyclic dione 19 (125 mg, 0.49 mmol) was dissolved in dry ethyl acetate (500 mL) and irradiated in a Pyrex immersion well by a 125 W lamp (homemade) for 1.5 h under nitrogen at rt. After completion of the reaction (TLC monitoring), the solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (5% ethyl acetate/petroleum ether) to give 20 (80 mg, 80%) as a thick pale yellow liquid. IR (KBr) νmax: 3073, 2974, 1752, 1636, 924 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.82 (d, J = 11.4 Hz, 1H), 2.12 (d, J = 11.4 Hz, 1H) 2.15–2.4 (m, 6H), 2.55 (dd, J1 = J2 = 1.0 Hz, 1H), 2.7 (dd, J1 = J2 = 1.0 Hz, 1H), 2.8–3.1 (m, 2H), 5.0–5.2 (m, 4H), 5.6–5.9 (m, 2H) ppm; 13C NMR (100.6 MHz, CDCl3) δ 33.7, 35.3, 35.6, 39.7, 42.9, 44.0, 47.3, 47.8, 51.5, 60.2, 61.6, 118.5, 118.7, 132.9, 133.7, 212.5, 213.6 ppm; HRMS (Q-ToF ES+) m/z: calcd for C17H19O2Na, 277.1204; found, 277.1210 [M + Na]+.
Preparation of pentacyclic tetraallyldiol 21
To a freshly prepared solution of allylmagnesium bromide in ether was added an ethereal solution of pentacyclic dione 20 (300 mg, 1.18 mmol) in a dropwise manner over a period of 10–15 min at rt under nitrogen. After completion of the reaction (TLC monitoring, 10 h), the reaction was quenched with a saturated aqueous NH4Cl solution at 0 °C. Then, the aqueous layer was extracted by ethyl acetate (3 × 25 mL). The combined organic layer was washed with brine, and dried over anhydrous Na2SO4. After the removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (4% ethyl acetate/petroleum ether) to give 21 (280 mg, 70%) as a white solid. Mp: 158–160 °C; IR (KBr) νmax: 3309, 3055, 2977, 1265, 743, 705 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.3 (1/2 ABq, J1 = 10.9 Hz, J2 = 11.0 Hz, 2H), 1.96–2.19 (m, 6H), 2.23–2.46 (m, 8H) 5.08–5.10 (m, 8H), 5.9–6.1 (m, 4H) ppm; 13C NMR (100.6 MHz, CDCl3) δ 33.1, 35.2, 36.6, 38.1, 41.1, 42.2, 43.1, 44.2, 45.2, 49.0, 49.5, 55.5, 55.7, 78.9, 80.5, 116.71, 116.72, 117.4, 118.3, 134.1, 134.7, 136.1, 136.3 ppm; HRMS (Q-ToF ES+) m/z: calcd for C23H30O2Na, 361.2144; found, 361.2146 [M + Na]+.
Preparation of heptacyclic diol 22
To a solution of 21 (25 mg, 0.074 mmol) was added Grubbs’ 2nd generation catalyst (4 mg, 6 mol %) under argon at rt. After completion of the reaction (TLC monitoring, 8 h), the solvent was evaporated and the resulting residue was purified by silica gel column chromatography (25% ethyl acetate/petroleum ether) to give 22 (15 mg, 72%) as a white crystalline solid. Mp: 206–207 °C; IR (KBr) νmax: 3691, 3054, 2987, 2305, 1422, 1266 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.38 (1/2 AB q, J1 = J2 = 11.0 Hz, 2H), 1.79 (q, J = 1.8 Hz, 1H), 1.99–2.56 (m, 13H), 5.54–5.72 (m, 4H) ppm; 13C NMR (100.6 MHz, CDCl3) δ 31.0, 31.4, 32.4, 36.2, 37.1, 37.7, 44.2, 44.5, 45.1, 47.3, 49.6, 51.4, 58.6, 74.7, 75.0, 123.1, 124.1, 124.4, 126.3 ppm; HRMS (Q-ToF ES+) m/z: calcd for C19H22O2Na, 305.1517; found, 305.1523 [M + Na]+.
Preparation of hexacyclic diallyldiol 24
To a freshly prepared solution of allylmagnesium bromide (6 equiv) in ether was added the ethereal solution of hexacyclic dione 23 (200 mg, 0.88 mmol) in a dropwise manner over a period of 10–15 min under nitrogen at rt. After completion of the reaction (TLC monitoring, 8 h), the reaction mixture was quenched with saturated aqueous NH4Cl solution at 0 °C. Then, the aqueous layer was extracted by ethyl acetate (3 × 25 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the resulting residue was purified by silica-gel column chromatography (4% ethyl acetate/petroleum ether) to give 24 (232 mg, 82%) as a white crystalline solid. Mp: 177–178 °C; IR (KBr) νmax: 3339, 3054, 2985, 1422, 1265 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.38 (1/2 ABq, J1 = J2 = 10.3 Hz, 2H), 1.9–2.0 (m, 6H), 2.13–2.25 (m, 6H), 2.32 (d, J = 1.6 Hz, 2H), 5.10–5.17 (m, 4H), 5.91–6.02 (m, 4H) ppm; 13C NMR (100.6 MHz, CDCl3) δ 26.2, 34.7, 42.1, 43.4, 43.7, 48.8, 51.4, 78.3, 117.9, 128.2, 134.0 ppm; HRMS (Q-ToF ES+) m/z: calcd for C20H27O2, 311.2011; found, 311.2012 [M + H]+.
Preparation of hexacyclic diallyldiol 26
To a freshly prepared solution of allylmagnesium bromide (6 equiv) in ether was added the ethereal solution of hexacyclic dione 25 (250 mg, 1.12 mmol) in a dropwise manner over a period of 10–15 min under nitrogen at rt. After completion of the reaction (TLC monitoring, 8 h), the reaction mixture was quenched with saturated aqueous NH4Cl solution at 0 °C. Then, the aqueous layer was extracted with ethyl acetate (3 × 25 mL). The combined organic layer was washed with brine and collected over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the resulting residue was purified by silica-gel column chromatography (4% ethyl acetate/petroleum ether) to give 26 (292 mg, 85%) as a white crystalline solid. Mp: 191–192 °C; IR (KBr) νmax: 3339, 3055, 2961, 1439, 1265 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.90 (d, J = 10.8 Hz, 1H), 1.40 (d, J = 10.8 Hz, 1H), 2.02–2.07 (m, 2H), 2.26 (s, 2H), 2.34–2.39 (m, 4H), 2.73 (s, 2H), 4.84 (brs, 2H), 5.10–5.15 (m, 4H), 5.56 (dd, J1 = 10.7 Hz, J2 = 2.7 Hz, 2H), 5.89–6.01 (m, 4H) ppm; 13C NMR (100.6 MHz, CDCl3) δ 31.7, 42.1, 44.0, 48.6, 51.3, 54.1, 78.8, 118.2, 124.3, 124.4, 133.7 ppm; HRMS (Q-ToF ES+) m/z: calcd for C21H25O2, 309.1855; found, 309.1862 [M + H]+.
Supporting Information
| Supporting Information File 1: Copies of 1H, 13C NMR and HRMS spectra for all new compounds. | ||
| Format: PDF | Size: 836.0 KB | Download |
References
-
Marchand, A. P. Aldrichimica Acta 1995, 28, 95.
Return to citation in text: [1] [2] -
Marchand, A. P. In Advances in Theoretically Interesting Molecules; Thummel, R. P., Ed.; JAI: Greenwich, CT, 1989; Vol. 1, pp 357 ff.
Return to citation in text: [1] [2] -
Mehta, G.; Srikrishna, A. Chem. Rev. 1997, 97, 671. doi:10.1021/cr9403650
Return to citation in text: [1] [2] -
Fessner, W. D.; Sedelmeier, G.; Spurr, P. R.; Rihs, G.; Prinzbach, H. J. Am. Chem. Soc. 1987, 109, 4626. doi:10.1021/ja00249a029
Return to citation in text: [1] [2] -
Olah, G. Cage Hydrocarbons; John-Wiley and Sons: New York, 1990.
Return to citation in text: [1] -
Osawa, E.; Yonemitsu, O. Carbocyclic Caged Compounds; VCH: New York, 1992.
Return to citation in text: [1] -
Hargittai, I.; Hargittai, M. Symmetry through the Eyes of a Chemist; Wiley: New York, 1987.
Return to citation in text: [1] -
Hopf, H. Classics in Hydrocarbon Chemistry; Wiley-VCH: Weinheim, 2000.
Return to citation in text: [1] -
McGlinchey, M. J.; Hopf, H. Beilstein J. Org. Chem. 2011, 7, 222. doi:10.3762/bjoc.7.30
Return to citation in text: [1] -
Van der Schyf, C. J.; Dekker, T. G.; Snyckers, F. O. Arch. Pharm. 1986, 319, 409. doi:10.1002/ardp.19863190506
Return to citation in text: [1] -
Van der Schyf, C. J.; Dekker, T. G.; Snyckers, F. Q.; Squier, G. J.; Coetzee, W. A.; Van der Walt, J. J.; Fourie, T. G.; Liebenberg, W. Polycyclic compounds and pharmaceutical compositions thereof. Eur. Pat. EP0312245, April 19, 1989.
Return to citation in text: [1] -
Oliver, D. W.; Malan, S. F. Med. Chem. Res. 2008, 17, 137. doi:10.1007/s00044-007-9044-5
Return to citation in text: [1] -
Marchand, A. P.; Kruger, H. G.; Power, E. D.; Segal, C. Kem. Ind. 2002, 51, 51.
Return to citation in text: [1] -
Marchand, A. P.; Dave, P. R.; Rajapaksa, D.; Arney, B. E., Jr.; Flippen-Anderson, J. L.; Gilardi, R.; George, C. J. Org. Chem. 1989, 54, 1769. doi:10.1021/jo00268a056
Return to citation in text: [1] -
Marchand, A. P.; Arney, B. E., Jr.; Dave, P. R. J. Org. Chem. 1988, 53, 443. doi:10.1021/jo00237a046
Return to citation in text: [1] -
Govender, T.; Hariprakasha, H. K.; Kruger, H. G.; Marchand, A. P. Tetrahedron: Asymmetry 2003, 14, 1553. doi:10.1016/S0957-4166(03)00272-6
Return to citation in text: [1] [2] -
Marchand, A. P.; Chong, H.-S.; Ganguly, B. Tetrahedron: Asymmetry 1999, 10, 4695. doi:10.1016/S0957-4166(99)00549-2
Return to citation in text: [1] -
Chamot, E.; Paquette, L. A. J. Org. Chem. 1978, 43, 4527. doi:10.1021/jo00417a031
Return to citation in text: [1] -
Paquette, L. A.; Beckley, R. S. J. Am. Chem. Soc. 1975, 97, 1084. doi:10.1021/ja00838a023
Return to citation in text: [1] -
Maier, G.; Pfriem, S.; Schäfer, U.; Matusch, R. Angew. Chem. 1978, 90, 552. doi:10.1002/ange.19780900714
Angew. Chem., Int. Ed. Engl. 1978, 14, 520. doi:10.1002/anie.197805201
Return to citation in text: [1] -
Katz, T. J.; Acton, N. J. Am. Chem. Soc. 1973, 95, 2738. doi:10.1021/ja00789a084
Return to citation in text: [1] -
McNichols, A. T.; Stang, P. J. Synlett 1992, 971. doi:10.1055/s-1992-21549
Return to citation in text: [1] -
Eaton, P. E.; Cole, T. W. J. Am. Chem. Soc. 1964, 86, 962. doi:10.1021/ja01059a072
Return to citation in text: [1] -
Mehta, G.; Padma, S. J. Am. Chem. Soc. 1987, 109, 7230. doi:10.1021/ja00257a076
Return to citation in text: [1] -
Ternansky, R. J.; Balogh, D. W.; Paquette, L. A. J. Am. Chem. Soc. 1982, 104, 4503. doi:10.1021/ja00380a040
Return to citation in text: [1] -
Prakash, G. K. S.; Krishnamurthy, V. V.; Herges, R.; Bau, R.; Yuan, H.; Olah, G. A.; Fessner, W.-D.; Prinzbach, H. J. Am. Chem. Soc. 1986, 108, 836. doi:10.1021/ja00264a046
Return to citation in text: [1] -
Mehta, G.; Reddy, S. H. K. Angew. Chem. 1993, 105, 1230. doi:10.1002/ange.19931050825
Angew. Chem., Int. Ed. Engl. 1993, 32, 1160. doi:10.1002/anie.199311601
Return to citation in text: [1] -
Kotha, S.; Dipak, M. K. Chem. – Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366
Return to citation in text: [1] [2] [3] [4] [5] -
Fieser, L. F.; Campbell, W. P.; Fry, E. M. J. Am. Chem. Soc. 1939, 61, 2206. doi:10.1021/ja01877a068
Return to citation in text: [1] -
Martin Castro, A. M. Chem. Rev. 2004, 104, 2939. doi:10.1021/cr020703u
Return to citation in text: [1] -
Griesbeck, A. G. Tetrahedron Lett. 1988, 29, 3477. doi:10.1016/0040-4039(88)85194-3
Return to citation in text: [1] -
Grubbs, R. H. Tetrahedron 2004, 60, 7117. doi:10.1016/j.tet.2004.05.124
Return to citation in text: [1] -
Deiters, A.; Martin, S. F. Chem. Rev. 2004, 104, 2199. doi:10.1021/cr0200872
Return to citation in text: [1] -
McReynolds, M. D.; Dougherty, J. M.; Hanson, P. R. Chem. Rev. 2004, 104, 2239. doi:10.1021/cr020109k
Return to citation in text: [1] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem. 2003, 115, 4740. doi:10.1002/ange.200300576
Angew. Chem., Int. Ed. 2003, 42, 4592. doi:10.1002/anie.200300576
Return to citation in text: [1] -
Kotha, S.; Sreenivasachary, N. Indian J. Chem., Sect. B 2001, 40, 763.
Return to citation in text: [1] -
Kotha, S.; Lahiri, K. Synlett 2007, 2767. doi:10.1055/s-2007-990954
Return to citation in text: [1] -
Valmiki Ramayana; Geeta Press: Gorakhpur, Uttar Pradesh, India; Sloka 6-7, p. 1425.
Return to citation in text: [1] -
Chitra Ramayana by Ramachandra Madhwa Mahishi, Illustrated by Balasaheb Pandit Pant Pratinidhi, 1916.
Return to citation in text: [1] -
Nickon, A.; Silversmith, E. F. Organic Chemistry. The Name Game Modern Coined Terms and Their Origins; Pergamon Press, 1987.
Return to citation in text: [1] -
Since the shape of molecule 22 resembles the ancient flying machine “Pushpak Viman” we coined the name ‘pushpakenediol’ for this dihydroxy caged system.
Return to citation in text: [1] -
Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064. doi:10.1002/asia.200700072
Return to citation in text: [1] [2] -
Mehta, G.; Uma, R. Acc. Chem. Res. 2000, 33, 278. doi:10.1021/ar990123s
Return to citation in text: [1]
| 28. | Kotha, S.; Dipak, M. K. Chem. – Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366 |
| 16. | Govender, T.; Hariprakasha, H. K.; Kruger, H. G.; Marchand, A. P. Tetrahedron: Asymmetry 2003, 14, 1553. doi:10.1016/S0957-4166(03)00272-6 |
| 1. | Marchand, A. P. Aldrichimica Acta 1995, 28, 95. |
| 2. | Marchand, A. P. In Advances in Theoretically Interesting Molecules; Thummel, R. P., Ed.; JAI: Greenwich, CT, 1989; Vol. 1, pp 357 ff. |
| 3. | Mehta, G.; Srikrishna, A. Chem. Rev. 1997, 97, 671. doi:10.1021/cr9403650 |
| 4. | Fessner, W. D.; Sedelmeier, G.; Spurr, P. R.; Rihs, G.; Prinzbach, H. J. Am. Chem. Soc. 1987, 109, 4626. doi:10.1021/ja00249a029 |
| 5. | Olah, G. Cage Hydrocarbons; John-Wiley and Sons: New York, 1990. |
| 6. | Osawa, E.; Yonemitsu, O. Carbocyclic Caged Compounds; VCH: New York, 1992. |
| 7. | Hargittai, I.; Hargittai, M. Symmetry through the Eyes of a Chemist; Wiley: New York, 1987. |
| 8. | Hopf, H. Classics in Hydrocarbon Chemistry; Wiley-VCH: Weinheim, 2000. |
| 9. | McGlinchey, M. J.; Hopf, H. Beilstein J. Org. Chem. 2011, 7, 222. doi:10.3762/bjoc.7.30 |
| 1. | Marchand, A. P. Aldrichimica Acta 1995, 28, 95. |
| 2. | Marchand, A. P. In Advances in Theoretically Interesting Molecules; Thummel, R. P., Ed.; JAI: Greenwich, CT, 1989; Vol. 1, pp 357 ff. |
| 27. |
Mehta, G.; Reddy, S. H. K. Angew. Chem. 1993, 105, 1230. doi:10.1002/ange.19931050825
Angew. Chem., Int. Ed. Engl. 1993, 32, 1160. doi:10.1002/anie.199311601 |
| 16. | Govender, T.; Hariprakasha, H. K.; Kruger, H. G.; Marchand, A. P. Tetrahedron: Asymmetry 2003, 14, 1553. doi:10.1016/S0957-4166(03)00272-6 |
| 17. | Marchand, A. P.; Chong, H.-S.; Ganguly, B. Tetrahedron: Asymmetry 1999, 10, 4695. doi:10.1016/S0957-4166(99)00549-2 |
| 28. | Kotha, S.; Dipak, M. K. Chem. – Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366 |
| 13. | Marchand, A. P.; Kruger, H. G.; Power, E. D.; Segal, C. Kem. Ind. 2002, 51, 51. |
| 14. | Marchand, A. P.; Dave, P. R.; Rajapaksa, D.; Arney, B. E., Jr.; Flippen-Anderson, J. L.; Gilardi, R.; George, C. J. Org. Chem. 1989, 54, 1769. doi:10.1021/jo00268a056 |
| 15. | Marchand, A. P.; Arney, B. E., Jr.; Dave, P. R. J. Org. Chem. 1988, 53, 443. doi:10.1021/jo00237a046 |
| 24. | Mehta, G.; Padma, S. J. Am. Chem. Soc. 1987, 109, 7230. doi:10.1021/ja00257a076 |
| 10. | Van der Schyf, C. J.; Dekker, T. G.; Snyckers, F. O. Arch. Pharm. 1986, 319, 409. doi:10.1002/ardp.19863190506 |
| 11. | Van der Schyf, C. J.; Dekker, T. G.; Snyckers, F. Q.; Squier, G. J.; Coetzee, W. A.; Van der Walt, J. J.; Fourie, T. G.; Liebenberg, W. Polycyclic compounds and pharmaceutical compositions thereof. Eur. Pat. EP0312245, April 19, 1989. |
| 12. | Oliver, D. W.; Malan, S. F. Med. Chem. Res. 2008, 17, 137. doi:10.1007/s00044-007-9044-5 |
| 25. | Ternansky, R. J.; Balogh, D. W.; Paquette, L. A. J. Am. Chem. Soc. 1982, 104, 4503. doi:10.1021/ja00380a040 |
| 26. | Prakash, G. K. S.; Krishnamurthy, V. V.; Herges, R.; Bau, R.; Yuan, H.; Olah, G. A.; Fessner, W.-D.; Prinzbach, H. J. Am. Chem. Soc. 1986, 108, 836. doi:10.1021/ja00264a046 |
| 20. |
Maier, G.; Pfriem, S.; Schäfer, U.; Matusch, R. Angew. Chem. 1978, 90, 552. doi:10.1002/ange.19780900714
Angew. Chem., Int. Ed. Engl. 1978, 14, 520. doi:10.1002/anie.197805201 |
| 19. | Paquette, L. A.; Beckley, R. S. J. Am. Chem. Soc. 1975, 97, 1084. doi:10.1021/ja00838a023 |
| 23. | Eaton, P. E.; Cole, T. W. J. Am. Chem. Soc. 1964, 86, 962. doi:10.1021/ja01059a072 |
| 18. | Chamot, E.; Paquette, L. A. J. Org. Chem. 1978, 43, 4527. doi:10.1021/jo00417a031 |
| 3. | Mehta, G.; Srikrishna, A. Chem. Rev. 1997, 97, 671. doi:10.1021/cr9403650 |
| 4. | Fessner, W. D.; Sedelmeier, G.; Spurr, P. R.; Rihs, G.; Prinzbach, H. J. Am. Chem. Soc. 1987, 109, 4626. doi:10.1021/ja00249a029 |
| 21. | Katz, T. J.; Acton, N. J. Am. Chem. Soc. 1973, 95, 2738. doi:10.1021/ja00789a084 |
| 31. | Griesbeck, A. G. Tetrahedron Lett. 1988, 29, 3477. doi:10.1016/0040-4039(88)85194-3 |
| 29. | Fieser, L. F.; Campbell, W. P.; Fry, E. M. J. Am. Chem. Soc. 1939, 61, 2206. doi:10.1021/ja01877a068 |
| 30. | Martin Castro, A. M. Chem. Rev. 2004, 104, 2939. doi:10.1021/cr020703u |
| 28. | Kotha, S.; Dipak, M. K. Chem. – Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366 |
| 42. | Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064. doi:10.1002/asia.200700072 |
| 28. | Kotha, S.; Dipak, M. K. Chem. – Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366 |
| 28. | Kotha, S.; Dipak, M. K. Chem. – Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366 |
| 41. | Since the shape of molecule 22 resembles the ancient flying machine “Pushpak Viman” we coined the name ‘pushpakenediol’ for this dihydroxy caged system. |
| 42. | Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064. doi:10.1002/asia.200700072 |
| 32. | Grubbs, R. H. Tetrahedron 2004, 60, 7117. doi:10.1016/j.tet.2004.05.124 |
| 33. | Deiters, A.; Martin, S. F. Chem. Rev. 2004, 104, 2199. doi:10.1021/cr0200872 |
| 34. | McReynolds, M. D.; Dougherty, J. M.; Hanson, P. R. Chem. Rev. 2004, 104, 2239. doi:10.1021/cr020109k |
| 35. |
Schrock, R. R.; Hoveyda, A. H. Angew. Chem. 2003, 115, 4740. doi:10.1002/ange.200300576
Angew. Chem., Int. Ed. 2003, 42, 4592. doi:10.1002/anie.200300576 |
| 36. | Kotha, S.; Sreenivasachary, N. Indian J. Chem., Sect. B 2001, 40, 763. |
| 37. | Kotha, S.; Lahiri, K. Synlett 2007, 2767. doi:10.1055/s-2007-990954 |
| 38. | Valmiki Ramayana; Geeta Press: Gorakhpur, Uttar Pradesh, India; Sloka 6-7, p. 1425. |
| 39. | Chitra Ramayana by Ramachandra Madhwa Mahishi, Illustrated by Balasaheb Pandit Pant Pratinidhi, 1916. |
| 40. | Nickon, A.; Silversmith, E. F. Organic Chemistry. The Name Game Modern Coined Terms and Their Origins; Pergamon Press, 1987. |
© 2014 Kotha and Dipak; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)