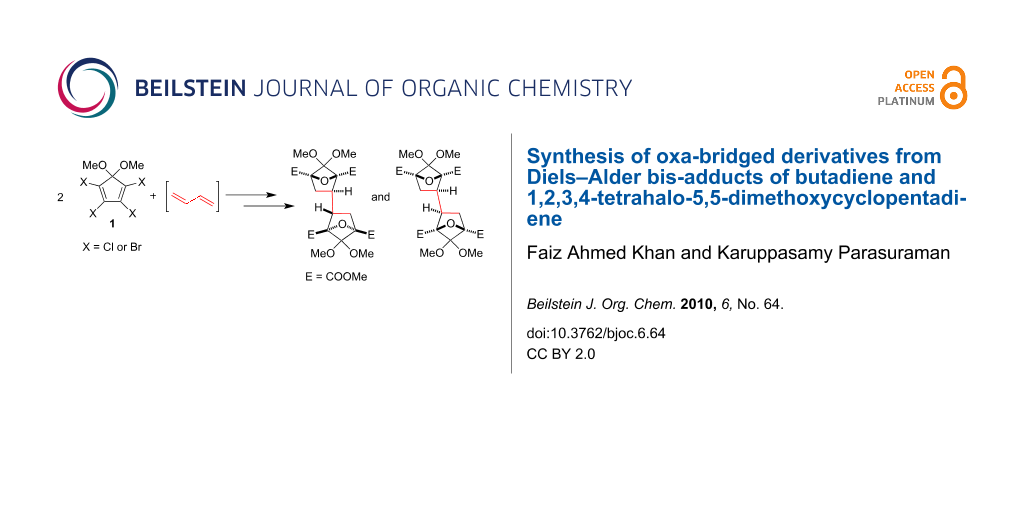
Abstract
Bis-adducts of 1,2,3,4-tetrahalo-5,5-dimethoxycyclopentadiene and 1,3-butadiene, generated in situ from 3-sulfolene, have been synthesized in excellent yield. Ruthenium catalyzed oxidation of the bis-adducts followed by a one-pot transformation of the resulting α-diketone furnished oxa-bridged compounds. Unambiguous stereochemical assignments of both diastereomeric series are reported.

Graphical Abstract
Introduction
3-Sulfolene is a nonflammable, nontoxic, nonhygroscopic and stable crystalline solid and is a convenient equivalent for gaseous 1,3-butadiene [1-3] and is commonly used for in situ generation of 1,3-butadiene as the diene component in Diels–Alder reactions. We and other groups have demonstrated the utility of cyclic dienes for the synthesis of 2:1 Diels–Alder bis-adducts with 1,2,3,4-tetrahalo-5,5-dimethoxycyclopentadiene 1 [4-7]. In the case of cyclic dienes (or trienes) such as cyclohexa-1,4-diene and cycloheptatriene, endo-syn-endo diastereomer 2 is formed exclusively, whilst cyclopentadiene and furan yield solely endo-anti-endo diastereomer 3 (Scheme 1). In continuation of our interest in the Diels–Alder bis-adducts of 1,2,3,4-tetrahalo-5,5-dimethoxycyclopentadienes 1 and their applications [8-14], we envisaged employing 1,3-butadiene as bis-dienophile component. Herein we report the synthesis of bis-adducts of 1,2,3,4-tetrahalo-5,5-dimethoxycyclopentadiene and butadiene followed by their transformation to oxa-bridged compounds. The stereochemistry of the diastereomeric products was also unequivocally established.
![[1860-5397-6-64-i1]](/bjoc/content/inline/1860-5397-6-64-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Diels–Alder bis-adducts of 1 with cyclic dienes.
Scheme 1: Diels–Alder bis-adducts of 1 with cyclic dienes.
We were interested in exploring the previously overlooked stereochemical outcome of the Diels–Alder reaction between 1a and 1,3-butadiene [15,16]. The bis-adduct obtained from 1a and gaseous 1,3-butadiene was previously assigned as “endo, exo-bis(7,7-dimethoxy-1,2,3,4-tetrachloronorborn-2-en-5-yl)” [16]. In our reinvestigation we used 3-sulfolene as a 1,3-butadiene source to prepare both the mono- and bis-adducts. The two diastereomeric bis-adducts were separated and the relative stereochemistry was established by single crystal X-ray diffraction and 1H NMR spectroscopy. The bis-adducts were further transformed into bis-diketones by means of supported ruthenium catalyzed oxidation. Finally, the two diastereomeric norbornyl α-diketones from the chloro as well as the bromo series were each converted to the corresponding oxa-bridged compounds [7].
Results and Discussion
For the preparation of the 2:1 adducts, 2 equivalents of 1,2,3,4-tetrachlorodimethoxycyclopentadiene 1a and one equivalent of 3-sulfolene were heated at 140–150 °C for 69 h in a sealed tube. The reaction mixture was purified by silica gel chromatography to afford the mono-adduct 4 in 7% yield as an inseparable mixture of endo and exo isomers [16] (endo:exo = 90:10, as determined by 1H NMR spectroscopy) and the two diastereomeric bis-adducts 5 and 6 as a 1:1 mixture in 92% yield (Scheme 2).
![[1860-5397-6-64-i2]](/bjoc/content/inline/1860-5397-6-64-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Diels–Alder reaction of 1a with 3-sulfolene.
Scheme 2: Diels–Alder reaction of 1a with 3-sulfolene.
The assignment for the exo-isomer 4 is based on the H5-endo methine signal at 2.48 ppm which appears as a triplet of doublets. The corresponding H5-exo methine proton for endo-isomer 4 appeared at 3.2 ppm. The bis-adducts 5 and 6 were successfully separated by preparative HPLC [17]. Adduct 5, a colourless crystalline compound with melting point 176–178 °C, displayed two singlets at 3.54 and 3.51 ppm for the methoxy groups, a multiplet at 2.45–2.42 ppm for two methine protons and another multiplet at 2.37–2.31 ppm for four methylene protons in its 1H NMR spectrum. In the 13C NMR spectrum, the methine carbon atoms appeared at 47.6 ppm, and the methylene carbon atoms at 41.4 ppm. By contrast, the diastereomer 6, a colorless solid with melting point 182–184 °C showed two singlets at 3.57 and 3.50 ppm for methoxy groups, a doublet of doublets at 2.96 ppm for methine protons and two doublets of doublets at 2.33 and 1.34 ppm for the methylene protons in its 1H NMR spectrum. In the 13C NMR spectrum of 6, the methine carbon atoms appeared at 43.7 ppm and the methylene carbons at 35.9 ppm.
The bis-adducts 5 and 6 were smoothly transformed to the corresponding bis-α-diketones 7 and 9 in excellent yield with a supported ruthenium catalyst (Ru-LDH) and NaIO4 as stoichiometric co-oxidant, a methodology developed in our laboratory [18,19]. Previously, we reported a smooth one-pot transformation of norbornyl α-diketones to the corresponding oxa-bridged derivatives [7], but our initial attempts to transform the bis-diketones 7 and 9 to bis-oxa-bridged compounds 8 and 10 using this strategy did not give the desired result. However, when the reaction was carried out in presence of the phase transfer catalyst TBHSO4 the bis-oxa-bridged compounds 8 and 10 were obtained (after esterification with diazomethane) in 31 and 37%, respectively (Scheme 3).
![[1860-5397-6-64-i3]](/bjoc/content/inline/1860-5397-6-64-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of bis-oxa-bridged compounds 8 and 10 from bis-adducts 5 and 6.
Scheme 3: Synthesis of bis-oxa-bridged compounds 8 and 10 from bis-adducts 5 and 6.
The relative stereochemistry in 8 was unambiguously established by the single crystal X-ray analysis (Figure 1) [20]. Working backwards, the structures of the adduct 5, the bis-diketone 7 were confirmed unequivocally.
![[1860-5397-6-64-1]](/bjoc/content/figures/1860-5397-6-64-1.png?scale=3.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP structure of 8 [50% probability thermal ellipsoids; some of the hydrogen atoms and a solvent molecule (acetonitrile) are omitted for clarity].
Figure 1: ORTEP structure of 8 [50% probability thermal ellipsoids; some of the hydrogen atoms and a solvent ...
We next turned our attention to the bromo analogue 1b in order to see if the overall yield of the bis-oxa-bridged derivatives 8 and 10 could be improved. We were also interested to see if any bromo derivative, corresponding to the diastereomer 6 in the chloro series, would furnish crystals suitable for X-ray analysis. The Diels–Alder reaction between 1,2,3,4-tetrabromo-5,5-dimethoxycyclopentadiene 1b and 3-sulfolene under the same experimental conditions as described for the chloro-analogue furnished mono-adduct 11 (endo:exo = 91:9) and bis-adducts 12 and 13 (Scheme 4). The bis-adducts 12 and 13 were separated by preparative HPLC.
![[1860-5397-6-64-i4]](/bjoc/content/inline/1860-5397-6-64-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Diels–Alder reaction of 1b with 3-sulfolene.
Scheme 4: Diels–Alder reaction of 1b with 3-sulfolene.
The bis-adducts 12 and 13 were converted in excellent yields to the corresponding bis-α-diketones 14 and 15 (Scheme 5). Bis-diketone 14 was treated first with alkaline H2O2 and then with additional NaOH (60 equiv) at 60 °C followed by esterification with diazomethane to obtain the oxa-bridged compound 8 in 42% yield. Bis-diketone 15 was transformed into 10 in 39% yield by a similar method. Unlike the bis-diketones in chloro series (7 and 9), which required a phase transfer reagent (TBHSO4), the bromo bis-diketones 14 and 15 underwent transformation to the bis-oxa-briged derivative 8 and 10 under the usual procedure previously reported from our laboratory [7] (Scheme 5). Although the yields in the final step were moderate (42 and 39%), this corresponds to 63–65% per oxa-bridge formed which is gratifying considering the number of intermediates involved and possible side reactions.
![[1860-5397-6-64-i5]](/bjoc/content/inline/1860-5397-6-64-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of bis-oxa-bridged compounds 8 and 10 from bis-diketones 14 and 15.
Scheme 5: Synthesis of bis-oxa-bridged compounds 8 and 10 from bis-diketones 14 and 15.
Unfortunately, neither 13 nor 15 gave crystals suitable for X-ray analysis. However, unambiguous assignment was possible from the diagnostic chemical shifts and coupling constants observed for methine (H5) and methylene (H6 and H6’) protons of bis-adducts 6 and 13 (Figure 2). The appearance of H5 at ~3 ppm with characteristic coupling constants of ~9 and ~4 Hz to H6 and H6’, respectively, unequivocally supports the assigned structures. These values are consistent with several endo-substituted derivatives (R = alkyl-like groups) reported by us [9] and others [21,22]. The observed selectivity is in agreement with the strong endo-selectivity displayed by diene 1.
![[1860-5397-6-64-2]](/bjoc/content/figures/1860-5397-6-64-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR chemical shifts (in parentheses) and coupling constants (J) for the three interacting protons (H5, H6, and H6’ ; for the sake of convenience, numbering sequence of mono-adducts is adopted) of the bis-adducts 6 and 13.
Figure 2: 1H NMR chemical shifts (in parentheses) and coupling constants (J) for the three interacting proton...
From the above results it is clear that the diastereomeric bis-adducts 5, 6 and 12, 13 are formed via endo-endo addition. The proposed transition states for the formation of bis-adducts are shown in Figure 3. The initial endo-mono adduct (4 or 11) gives rise to two possible endo-transition states leading to 5, 6 or 12, 13. The corresponding exo-transition states suffer from severe steric congestion due to the bulky R group and are consequently unfavorable. Similar steric considerations rule out the participation of an initially formed minor exo-mono adduct (4 or 11) to participate further in the reaction to give bis-adducts, thus ruling out the formation of diastereomers via exo-endo addition.
![[1860-5397-6-64-3]](/bjoc/content/figures/1860-5397-6-64-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Transition state models for the bis-adduct formation.
Figure 3: Transition state models for the bis-adduct formation.
Conclusion
In conclusion, we have demonstrated that the Diels–Alder reaction between 1 (diene component) and 1,3-butadiene (bis-dienophile component) proceeds via endo-endo addition mode to give a 1:1 mixture of diastereomeric bis-adducts. The diastereomeric bis-adducts were separated and transformed into bis-oxa-bridged compounds. The relative stereochemistry of the products was unambiguously established by single crystal X-ray diffraction and NMR spectroscopy.
Supporting Information
| Supporting Information File 1: General methods, experimental procedures and analytical data for new compounds. | ||
| Format: PDF | Size: 362.4 KB | Download |
References
-
Fieser, L. F.; Fieser, M. Reagent for organic synthesis; Wiley: New York, 1969; Vol. 2, p 390.
Return to citation in text: [1] -
Sample, T. E., Jr..; Hatch, L. F. Org. Synth. 1988, 6, 454.
Return to citation in text: [1] -
Chou, T.-S.; Tso, H.-H. Org. Prep. Proced. Int. 1989, 21, 259–296.
Return to citation in text: [1] -
Forman, M. A.; Dailey, W. P. J. Org. Chem. 1993, 58, 1501–1507. doi:10.1021/jo00058a035
Return to citation in text: [1] -
Garcia, J. G.; Fronczek, F. R.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3289–3292. doi:10.1016/S0040-4039(00)92688-1
Return to citation in text: [1] -
Garcia, J. G.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3293–3296. doi:10.1016/S0040-4039(00)92689-3
Return to citation in text: [1] -
Khan, F. A.; Dash, J.; Sudheer, Ch.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385
Return to citation in text: [1] [2] [3] [4] -
Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424–2425. doi:10.1021/ja017371f
Return to citation in text: [1] -
Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, Ch. J. Org. Chem. 2002, 67, 3783–3787. doi:10.1021/jo025521e
Return to citation in text: [1] [2] -
Khan, F. A.; Dash, J. J. Org. Chem. 2003, 68, 4556–4559. doi:10.1021/jo034023i
Return to citation in text: [1] -
Khan, F. A.; Satapathy, R.; Dash, J.; Savitha, G. J. Org. Chem. 2004, 69, 5295–5301. doi:10.1021/jo049615v
Return to citation in text: [1] -
Khan, F. A.; Rout, B. Tetrahedron Lett. 2006, 47, 5251–5253. doi:10.1016/j.tetlet.2006.05.156
Return to citation in text: [1] -
Khan, F. A.; Rout, B. J. Org. Chem. 2007, 72, 7011–7013. doi:10.1021/jo0710127
Return to citation in text: [1] -
Khan, F. A.; Parasuraman, K.; Sadhu, K. K. Chem. Commun. 2009, 2399–2401. doi:10.1039/b820479a
Return to citation in text: [1] -
Peri, C. A. Gazz. Chim. Ital. 1955, 85, 1118.
(Chem. Abstr. 1956, 50, 10013).
Return to citation in text: [1] -
Nigmatova, V. B.; Zaitsev, Y. V.; Anfilogova, S. N.; Pekhk, T. I.; Belikova, N. A. Russ. J. Org. Chem. 1994, 30, 727–732.
Return to citation in text: [1] [2] [3] -
JAI LC-908W preparative HPLC equipped with a JAIGEL-OA4100 column (Japan Analytical Industry Co. Ltd.).
Return to citation in text: [1] -
Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558–9559. doi:10.1021/ja001956c
Return to citation in text: [1] -
Khan, F. A.; Sahu, N. J. Catal. 2005, 231, 438–442. doi:10.1016/j.jcat.2005.02.001
Return to citation in text: [1] -
Crystal data for 8: colorless crystal (recrystallized from acetonitrile solution). C22 H30 O14 2(C2 N), M = 594.53, 0.18 x 0.15 x 0.13 mm3, Triclinic, space group P-1 with a = 8.007(3) Å, b = 8.588(3) Å, c = 11.639(4) Å, α = 97.274(6)°, β = 98.309(6)°, γ = 110.118(6)°, V = 730.1(5) Å3, T = 100(2) K, R1 = 0.0786, wR2 = 0.2155 on observed data, z = 1, Dcalcd = 1.352 g∙cm−3, F(000) = 274, Absorption coefficient = 0.111 mm−1, λ = 0.71073 Å. The largest difference peak and hole = 0.515 and −0.352 eÅ−3, respectively. CCDC: 763534 contain the supplementary crystallographic data for the compounds 8. This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or mail to: deposit@ccdc.cam.ac.uk.
Return to citation in text: [1] -
Veliev, M. G.; Chalabieva, A. Z.; Shatirova, M. I.; Mamedov, E. Sh.; Mamedov, I. M. Russ. J. Org. Chem. 2001, 37, 223–229. doi:10.1023/A:1012326912373
Return to citation in text: [1] -
Veliev, M. G.; Chalabieva, A. Z.; Mamedova, A. F. Russ. J. Org. Chem. 2009, 45, 650–659.
Return to citation in text: [1]
| 1. | Fieser, L. F.; Fieser, M. Reagent for organic synthesis; Wiley: New York, 1969; Vol. 2, p 390. |
| 2. | Sample, T. E., Jr..; Hatch, L. F. Org. Synth. 1988, 6, 454. |
| 3. | Chou, T.-S.; Tso, H.-H. Org. Prep. Proced. Int. 1989, 21, 259–296. |
| 16. | Nigmatova, V. B.; Zaitsev, Y. V.; Anfilogova, S. N.; Pekhk, T. I.; Belikova, N. A. Russ. J. Org. Chem. 1994, 30, 727–732. |
| 15. |
Peri, C. A. Gazz. Chim. Ital. 1955, 85, 1118.
(Chem. Abstr. 1956, 50, 10013). |
| 16. | Nigmatova, V. B.; Zaitsev, Y. V.; Anfilogova, S. N.; Pekhk, T. I.; Belikova, N. A. Russ. J. Org. Chem. 1994, 30, 727–732. |
| 8. | Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424–2425. doi:10.1021/ja017371f |
| 9. | Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, Ch. J. Org. Chem. 2002, 67, 3783–3787. doi:10.1021/jo025521e |
| 10. | Khan, F. A.; Dash, J. J. Org. Chem. 2003, 68, 4556–4559. doi:10.1021/jo034023i |
| 11. | Khan, F. A.; Satapathy, R.; Dash, J.; Savitha, G. J. Org. Chem. 2004, 69, 5295–5301. doi:10.1021/jo049615v |
| 12. | Khan, F. A.; Rout, B. Tetrahedron Lett. 2006, 47, 5251–5253. doi:10.1016/j.tetlet.2006.05.156 |
| 13. | Khan, F. A.; Rout, B. J. Org. Chem. 2007, 72, 7011–7013. doi:10.1021/jo0710127 |
| 14. | Khan, F. A.; Parasuraman, K.; Sadhu, K. K. Chem. Commun. 2009, 2399–2401. doi:10.1039/b820479a |
| 9. | Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, Ch. J. Org. Chem. 2002, 67, 3783–3787. doi:10.1021/jo025521e |
| 4. | Forman, M. A.; Dailey, W. P. J. Org. Chem. 1993, 58, 1501–1507. doi:10.1021/jo00058a035 |
| 5. | Garcia, J. G.; Fronczek, F. R.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3289–3292. doi:10.1016/S0040-4039(00)92688-1 |
| 6. | Garcia, J. G.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3293–3296. doi:10.1016/S0040-4039(00)92689-3 |
| 7. | Khan, F. A.; Dash, J.; Sudheer, Ch.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385 |
| 21. | Veliev, M. G.; Chalabieva, A. Z.; Shatirova, M. I.; Mamedov, E. Sh.; Mamedov, I. M. Russ. J. Org. Chem. 2001, 37, 223–229. doi:10.1023/A:1012326912373 |
| 22. | Veliev, M. G.; Chalabieva, A. Z.; Mamedova, A. F. Russ. J. Org. Chem. 2009, 45, 650–659. |
| 18. | Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558–9559. doi:10.1021/ja001956c |
| 19. | Khan, F. A.; Sahu, N. J. Catal. 2005, 231, 438–442. doi:10.1016/j.jcat.2005.02.001 |
| 20. | Crystal data for 8: colorless crystal (recrystallized from acetonitrile solution). C22 H30 O14 2(C2 N), M = 594.53, 0.18 x 0.15 x 0.13 mm3, Triclinic, space group P-1 with a = 8.007(3) Å, b = 8.588(3) Å, c = 11.639(4) Å, α = 97.274(6)°, β = 98.309(6)°, γ = 110.118(6)°, V = 730.1(5) Å3, T = 100(2) K, R1 = 0.0786, wR2 = 0.2155 on observed data, z = 1, Dcalcd = 1.352 g∙cm−3, F(000) = 274, Absorption coefficient = 0.111 mm−1, λ = 0.71073 Å. The largest difference peak and hole = 0.515 and −0.352 eÅ−3, respectively. CCDC: 763534 contain the supplementary crystallographic data for the compounds 8. This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or mail to: deposit@ccdc.cam.ac.uk. |
| 17. | JAI LC-908W preparative HPLC equipped with a JAIGEL-OA4100 column (Japan Analytical Industry Co. Ltd.). |
| 7. | Khan, F. A.; Dash, J.; Sudheer, Ch.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385 |
| 16. | Nigmatova, V. B.; Zaitsev, Y. V.; Anfilogova, S. N.; Pekhk, T. I.; Belikova, N. A. Russ. J. Org. Chem. 1994, 30, 727–732. |
| 7. | Khan, F. A.; Dash, J.; Sudheer, Ch.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385 |
| 7. | Khan, F. A.; Dash, J.; Sudheer, Ch.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385 |
© 2010 Khan and Parasuraman; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)