Abstract
4-Aryl-4-oxoesters undergo facile reduction of both the keto and the ester groups with methanolic NaBH4 at room temperature to yield the corresponding 1-aryl-1,4-butanediols whereas 4-alkyl-4-oxoesters furnish the corresponding 1,4-butanolides via selective reduction of the keto moiety. Results of a detailed and systematic investigation of the reaction are described.

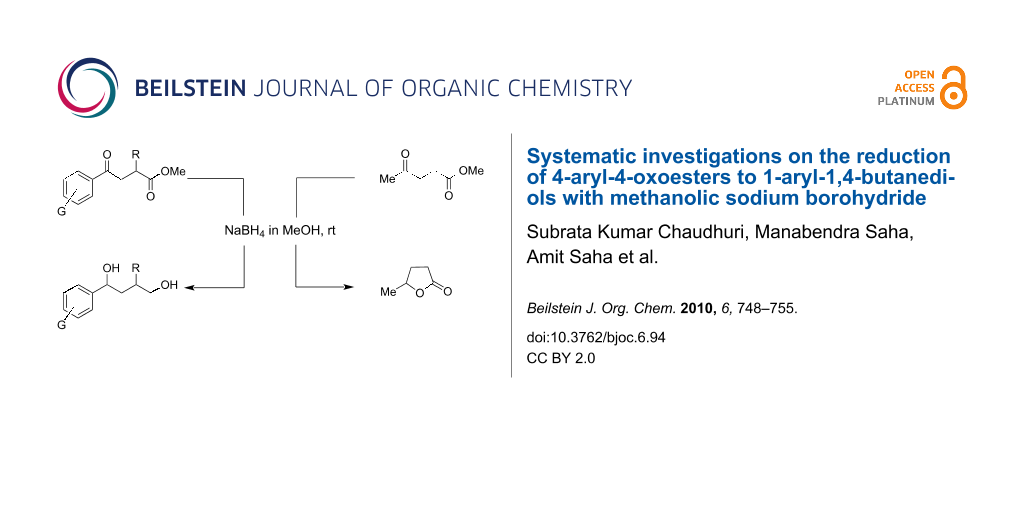
Graphical Abstract
Introduction
Chemoselective reductions of aldehydes, ketones and imines are generally accomplished using NaBH4 in methanol where other reducible functional groups, e.g. esters, nitro, nitriles, etc., remain unaffected [1-10]. Although it has been reported that some aliphatic and aromatic esters have been reduced with a large excess of sodium or other metal borohydrides [11,12], often in higher boiling solvents [13] and in combination with various additives [14,15] including at a cationic micellar surface [16], selective reduction of the keto group in oxoesters has been accomplished using potassium borohydride in refluxing ethanol [17] where the product distribution critically depends on the relative proportions of substrate and reagent. Despite the occurrence of several recent reports of borohydride-mediated reduction of the ester moiety in α-oxo- [18,19] and β-oxoesters [20], sodium borohydride in various alcoholic solvents, often in the presence of additives [21], has been judiciously utilized [22] for the chemoselective reduction of the oxo-group, occasionally with subsequent transesterification and the formation of the alkoxy-modified β-hydroxyesters. γ-Oxoesters react chemoselectively with sodium borohydride to produce the corresponding γ-hydroxyesters [1,2,17,23-27] (sometimes in the form of γ-lactone) [24]. Following the above noted literature precedences [1,2,17,22-27] on the utility of NaBH4, we attempted to reduce 4-aryl-4-oxoesters with methanolic NaBH4 chemoselectively. Surprisingly, we found that 4-aryl-4-oxoesters underwent facile reduction of both the keto and the ester groups with methanolic NaBH4 at room temperature to yield the corresponding 1-aryl-1,4-butanediols whereas 4-alkyl-4-oxoesters furnished the corresponding 1,4-butanolides via selective reduction of the keto moiety. These results, to the best of our knowledge, have no literature precedence. We describe herein our systematic investigations to elucidate the different parameters involved in these reactions and to establish their synthetic usefulness.
Results and Discussion
When, the γ-aryl-γ-ketoesters (1a–1f) were treated with methanolic NaBH4 (4 equiv) at room temperature (room temperature implies 30 °C throughout) both the oxo- and the alkoxycarbonyl moieties were reduced to give the diols (2a–2f), as shown in Scheme 1.
![[1860-5397-6-94-i1]](/bjoc/content/inline/1860-5397-6-94-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Facile reduction of γ-aryl-γ-ketoesters to the corresponding diols with methanolic NaBH4 at room temperature.
Scheme 1: Facile reduction of γ-aryl-γ-ketoesters to the corresponding diols with methanolic NaBH4 at room te...
γ-Aryl-α,β-unsaturated-γ-ketoesters (1g and 1h), on similar treatment, furnished the saturated diols (2a and 2b) by the reduction of both the keto and the ester groups along with complete hydrogenation of the double bond (Scheme 2).
![[1860-5397-6-94-i2]](/bjoc/content/inline/1860-5397-6-94-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Facile reduction of γ-aryl-α,β-unsaturated-γ-ketoesters to the diols with methanolic NaBH4 at room temperature.
Scheme 2: Facile reduction of γ-aryl-α,β-unsaturated-γ-ketoesters to the diols with methanolic NaBH4 at room ...
Detailed results are shown in Table 1.
Table 1: Reduction of 4-aryl-4-oxoesters (saturated and α,β-unsaturated) with NaBH4 in MeOH at room temperature (30 °C).
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-94-i12.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-94-i13.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-94-i14.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-94-i15.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-94-i16.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-6-94-i17.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-6-94-i18.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-6-94-i19.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-6-94-i20.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-6-94-i21.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-6-94-i22.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-6-94-i23.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-6-94-i24.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-6-94-i25.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-6-94-i26.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-6-94-i27.svg?max-width=637&scale=1.0)
aYields refer to pure products, fully characterized spectroscopically (1H NMR, 300 MHz). References for known compounds are given in parenthesis after the respective yields.
At this point it is very interesting and important to note that only the oxo function of 4-alkyl-4-oxoester 3 was selectively reduced under the same conditions to yield lactone 4 without affecting the oxidation state of the alkoxycarbonyl moiety (Scheme 3).
![[1860-5397-6-94-i3]](/bjoc/content/inline/1860-5397-6-94-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Facile reduction of γ-alkyl-γ-ketoester to the corresponding lactone with methanolic NaBH4 at room temperature.
Scheme 3: Facile reduction of γ-alkyl-γ-ketoester to the corresponding lactone with methanolic NaBH4 at room ...
From the results obtained so far, it is obvious that NaBH4 in methanol can be efficiently used for the synthesis of 1-aryl-1,4-butanediols from the easily accessible 4-aryl-4-oxoesters (Table 1) instead of employing the more costly and hazardous LiAlH4 which also often gives rise to several non-identifiable by-products. Structurally varied 1-aryl-1,4-butanediols are of great synthetic value with immense applications in cationic polymerizations [34], as intermediates for the syntheses of important acyclic antiviral nucleosides [35] and cyclic ethers [36].
Substrate 5 also underwent similar transformation under more drastic conditions to give a mixture of diol 6 [37] and lactone 7 [38], as shown in Scheme 4. In this instance no reaction took place at room temperature even after 24 h which might be ascribed to the lower electrophilicity of both the oxo- and alkoxycarbonyl functionalities of 5 from both electronic and steric standpoints.
![[1860-5397-6-94-i4]](/bjoc/content/inline/1860-5397-6-94-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reduction of methyl o-benzoylbenzoate with methanolic NaBH4.
Scheme 4: Reduction of methyl o-benzoylbenzoate with methanolic NaBH4.
The des-keto ester 8, as expected, was totally unaffected (Scheme 5) and was recovered unchanged.
![[1860-5397-6-94-i5]](/bjoc/content/inline/1860-5397-6-94-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reluctance of ester 8 towards reduction with methanolic NaBH4 at room temperature.
Scheme 5: Reluctance of ester 8 towards reduction with methanolic NaBH4 at room temperature.
Therefore, it is clear that the presence of both the aryl moiety and the oxo-function at the γ-carbon with respect to ester functionality is essential to bring about reduction of ester group with NaBH4. No reduction occurred when the reactions were carried out in anhydrous ether in place of methanol, however, substrates 1a and 1b in the ethereal medium underwent transformations in the presence of various protic polar co-solvents with different product distributions depending upon the nature of the co-solvent (Table 2).
Table 2: Reactionsa of 1a and 1b with NaBH4 in anhydrous ether in the presence of protic polar co-solvents.
| Entry | SM | Co-solvent | Relative product distribution (%)b | |||
|---|---|---|---|---|---|---|
| Substrate | Lactone | Diol | Hydroxyester | |||
| 1 | 1a | MeOH | – | 37.1 | 62.9 | – |
| 2 | 1a | EtOH | – | 40.6 | 59.4 | – |
| 3 | 1a | t-BuOH | 5.8 | 94.2 | – | – |
| 4 | 1a | H2O | 86.1 | 2.1 | 11.8 | – |
| 5 | 1a | AcOH | 87.3 | 3.1 | 9.6 | – |
| 6 | 1b | MeOH | – | Trace | 99.0 | – |
| 7 | 1b | EtOH | – | 48.2 | 51.8 | – |
| 8 | 1b | t-BuOH | 60.4 | 18.1 | – | 21.5 |
| 9 | 1b | H2O | 48.7 | 15.4 | – | 35.9 |
| 10 | 1b | AcOH | 21.9 | 51.4 | – | 26.6 |
aNaBH4 (4 equiv) in Et2O, co-solvent (2 equiv), 30 °C, 4 h. bDetermined by 300 MHz 1H NMR.
Compounds 1a, 3, acetophenone and butyrophenone were individually subjected to reduction in ether (Table 3) in the presence of MeOH (2 equiv) for a limited period of time (1 h). It was observed that the reduction of the keto group in the γ-oxoesters 1a and 3 (entries 1 and 2 in Table 3) with the formation of the lactones 9 and 4 as one of the products was much faster than the reduction of aryl alkyl ketones (entries 3 and 4 in Table 3). Therefore, formation of lactone as the intermediate might be crucial for more facile reduction of the keto moiety in case of γ-oxoesters (entries 1 and 2 in Table 3), which is not possible in the case of normal aryl alkyl ketones (entries 3 and 4 in Table 3). It is also interesting to note that although in both 1a and 3 the keto group was completely reduced, the relative proportion of the lactone (compared to hydroxyester) was much higher for 1a than for 3.
Table 3: Comparative studya on reduction of various oxo-groups.
| Entry | Substrate | Relative proportion (%)b of | ||
|---|---|---|---|---|
| Substrate | Reduced products | |||
| Lactone | γ-Hydroxyester | |||
| 1 | 1a | – | 62.5 | 37.5 |
| 2 | 3 | – | 32.1 | 67.9 |
| 3 | Acetophenone | 49.2 | 50.8 | |
| 4 | Butyrophenone | 61.0 | 39.0 | |
aNaBH4 (4 equiv) in Et2O, MeOH (2 equiv), 30 °C, 1 h. bDetermined by 300 MHz 1H NMR.
The intermediacy of lactone 9 [24] was also established by an independent route as outlined in Scheme 6.
![[1860-5397-6-94-i6]](/bjoc/content/inline/1860-5397-6-94-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Intermediacy of a lactone in the formation of diol.
Scheme 6: Intermediacy of a lactone in the formation of diol.
In order to prove the essentiality of the intermediacy of a lactone, compound 1g (with the keto and ester moieties kept far apart for lactonization due to trans-geometry of the olefinic linkage) was treated with NaBH4 (4 equiv) in methanol. However, this reaction unexpectedly led to the exclusive formation of 2a. With a smaller amount (2 equiv) of NaBH4 in methanol, compound 1g gave 9 and 2a in a ratio of 69:31(Scheme 7).
![[1860-5397-6-94-i7]](/bjoc/content/inline/1860-5397-6-94-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Diol formation from γ-aryl-α,β-unsaturated-γ-ketoester through the intermediacy of a saturated lactone during the reduction with methanolic NaBH4.
Scheme 7: Diol formation from γ-aryl-α,β-unsaturated-γ-ketoester through the intermediacy of a saturated lact...
It was presumed that the formation of 2a from 1g might occur through the initial reduction of the keto group with the formation of the γ-hydroxy-γ-aryl-α,β-unsaturated ester 10 [25]. In this connection it should be noted that when a limited amount of borohydride (1.2 equiv) was employed, we obtained the corresponding γ-hydroxy-trans-α,β-enoic ester 10 from 1g. γ-Hydroxy-α,β-acetylenic esters have been reported [26] to undergo conjugate reduction of the triple bond with NaBH4 at low temperature (−34 °C) to give the corresponding γ-hydroxy-α,β-alkenoic esters, where the conjugate reduction does not proceed beyond the double bond. However, we have observed conjugate reduction of γ-hydroxy-α,β-alkenoic esters with methanolic NaBH4 (4 equiv) at 30 °C during the transformation of 10 to 2a. Conjugate reduction here might be explained by the following plausible mechanistic scheme (Figure 1) where a mixed alkenyloxy alkoxy borohydride is initially formed by the reaction of 10 with sodium borohydride followed by conjugate reduction of olefinic linkage by intramolecular hydride attack to produce saturated 4-hydroxyester, which subsequently cyclizes to yield 9 and then further reduced to the diol 2a.
![[1860-5397-6-94-1]](/bjoc/content/figures/1860-5397-6-94-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Mechanistic rationale for diol formation during the reduction of a γ-aryl-α,β-unsaturated-γ-ketoester with methanolic NaBH4.
Figure 1: Mechanistic rationale for diol formation during the reduction of a γ-aryl-α,β-unsaturated-γ-ketoest...
This postulate is supported by the observation that the proposed intermediate 10 (independently synthesized from 11) is reduced to 2a by the present method (Scheme 8, dotted arrows denote the route proposed in Figure 1).
![[1860-5397-6-94-i8]](/bjoc/content/inline/1860-5397-6-94-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Intermediacy of γ-aryl-α,β-unsaturated-γ-hydroxyester during the reduction of γ-aryl-α,β-unsaturated-γ-ketoesters with methanolic NaBH4.
Scheme 8: Intermediacy of γ-aryl-α,β-unsaturated-γ-hydroxyester during the reduction of γ-aryl-α,β-unsaturate...
The fact that the reduction of the keto group occurs before the conjugate reduction of the olefinic linkage has also been established in this study. In the basic reaction medium produced by NaBH4, the –COOH group is converted to –COO−, and as a result the double bond is no longer electron-deficient. The conjugate reduction by the intramolecular nucleophilic attack of the hydride is therefore not feasible. As a consequence, the –OH and –COO− are too far apart to interact with each other. Therefore a single bond between the carbinol carbon and carboxylic acid moiety is impossible and hence no possibility of rotation, lactonization and subsequent reduction to diol 2a. For this reason the γ-keto-α,β-enoic acid 11 on treatment with 4 equiv of NaBH4 in methanol smoothly furnished 12 as the preponderant product without conjugate reduction and subsequent reductive functional group transformation.
When substrate 13 [39] (with vicinal anti-dibromo substituents to increase the rotational barrier of the single bond) was reacted with methanolic NaBH4 (4 equiv) at room temperature, a mixture of 9, 10 and 2a was obtained in a ratio of 44:15:41 (as determined by 300 MHz 1H NMR), as shown in Scheme 9.
![[1860-5397-6-94-i9]](/bjoc/content/inline/1860-5397-6-94-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reduction of γ-aryl-α,β-anti-dibromo-γ-ketoester with methanolic NaBH4.
Scheme 9: Reduction of γ-aryl-α,β-anti-dibromo-γ-ketoester with methanolic NaBH4.
Possibly, compound 13 was first reduced at the carbonyl function followed by concomitant dehydrobromination (under the basic reaction conditions), conjugate reduction at olefinic linkage, further dehydrobromination to 10 and subsequent conjugate reduction of 10 with the formation of 9 (as per the previous mechanistic scheme shown in Figure 1) and reduction of 9 to 2a. The formation of 10 from 13 has been confirmed by the isolation of 10 (as the major product) as the outcome of the reaction of 13 with a limited amount of NaBH4 (1.5 equiv), as shown in Scheme 10.
![[1860-5397-6-94-i10]](/bjoc/content/inline/1860-5397-6-94-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Intermediacy of γ-aryl-α,β-unsaturated-γ-hydroxyester during the reduction of γ-aryl-α,β-anti-dibromo-γ-ketoester with methanolic NaBH4.
Scheme 10: Intermediacy of γ-aryl-α,β-unsaturated-γ-hydroxyester during the reduction of γ-aryl-α,β-anti-dibro...
The crucial role of the lactone formation during the borohydride-mediated reduction of 4-aryl-4-oxoester to 1,4-diols was finally established (Scheme 11) when substrate 14 [40] (incapable of lactonization due to distal spatial disposition of the oxo- and methoxycarbonyl moieties imposed by the rigidity of the cyclopropane ring system) underwent selective reduction of the oxo-functionality only under refluxing conditions to yield 15. No significant reaction was observed at room temperature (monitored by TLC) even after 12 h.
![[1860-5397-6-94-i11]](/bjoc/content/inline/1860-5397-6-94-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Chemoselective reduction of keto group in the presence of ester moiety where structural rigidity prevents the formation of a lactone intermediate during the reduction of γ-aryl-γ-ketoester with methanolic NaBH4.
Scheme 11: Chemoselective reduction of keto group in the presence of ester moiety where structural rigidity pr...
From the investigations carried out so far, the intermediacy of a lactone during the NaBH4-mediated facile reduction of saturated and α,β-unsaturated-γ-aryl-γ-oxoesters to the corresponding saturated 1,4-butanediols has been firmly established. However, the reason for more facile reduction of the γ-aryl-lactones to diols and the relative reluctance of the γ-alkyl analogues is not yet clear.
Conclusion
From the above study, a novel method utilizing NaBH4 in methanol that can provide clean, cost-effective and facile access to differently substituted 1-aryl-1,4-butanediols in good yield and high purity from the easily accessible precursors has been developed. The results also indicate that caution should be exercised when methanolic sodium borohydride is used as a reagent [1,2,17,22-27] for the chemoselective reduction of the keto group of all types of γ-oxoesters.
Supporting Information
General experimental procedure for the NaBH4 reduction and the spectral data of the products are presented as supplementary data.
| Supporting Information File 1: Experimental. | ||
| Format: PDF | Size: 45.7 KB | Download |
Acknowledgements
The authors express sincere gratitude to Mr. N. Dutta of Indian Association for the Cultivation of Science, Kolkata, India for necessary assistance. Financial and infrastructural support from UGC-CAS programme in Chemistry, Jadavpur University, DST-PURSE programme and DST-FIST programme are also gratefully acknowledged.
References
-
Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, 3rd ed.; Part A and Part B; Plenum Press: New York, 1990.
Return to citation in text: [1] [2] [3] [4] -
House, H. O. Modern Synthetic Reactions; Benjamin/Cummings Publishing Company: MA, 1972.
Return to citation in text: [1] [2] [3] [4] -
Kim, J.; Bruning, J.; Park, K. E.; Lee, D. J.; Singaram, B. Org. Lett. 2009, 11, 4358–4361. doi:10.1021/ol901677b
Return to citation in text: [1] -
Alinezhad, H.; Tajbakhsh, M.; Zare, M. Synth. Commun. 2009, 39, 2907–2916. doi:10.1080/00397910802691882
Return to citation in text: [1] -
Sawada, T.; Ishii, H.; Ueda, T.; Aoki, J. Synth. Commun. 2009, 39, 3912–3923. doi:10.1080/00397910902883546
Return to citation in text: [1] -
Alinezhad, H.; Tajbakhsh, M.; Mahdavi, N. Synth. Commun. 2010, 40, 951–956. doi:10.1080/00397910903026731
Return to citation in text: [1] -
Cook, C.; Guinchard, X.; Liron, F.; Roulland, E. Org. Lett. 2010, 12, 744–747. doi:10.1021/ol902829e
Return to citation in text: [1] -
Watanabe, T.; Imaizumi, T.; Chinen, T.; Nagumo, Y.; Shibuya, M.; Usui, T.; Kanoh, N.; Iwabuchi, Y. Org. Lett. 2010, 12, 1040–1043. doi:10.1021/ol1000389
Return to citation in text: [1] -
McIver, A. L.; Deiters, A. Org. Lett. 2010, 12, 1288–1291. doi:10.1021/ol100177u
Return to citation in text: [1] -
Collins, J.; Rinner, U.; Moser, M.; Hudlicky, T.; Ghiviriga, I.; Romero, A. E.; Kornienko, A.; Ma, D.; Griffin, C.; Pandey, S. J. Org. Chem. 2010, 75, 3069–3084. doi:10.1021/jo1003136
Return to citation in text: [1] -
Brown, H. C.; Mead, E. J.; Subba Rao, B. C. J. Am. Chem. Soc. 1955, 77, 6209–6213. doi:10.1021/ja01628a044
Return to citation in text: [1] -
Brown, M. S.; Rapoport, H. J. Org. Chem. 1963, 28, 3261–3263. doi:10.1021/jo01046a538
Return to citation in text: [1] -
Soai, K.; Oyamada, H.; Ookawa, A. Synth. Commun. 1982, 12, 463–467. doi:10.1080/00397918208065953
Return to citation in text: [1] -
Yamakawa, T.; Masaki, M.; Nohira, H. Bull. Chem. Soc. Jpn. 1991, 64, 2730–2734. doi:10.1246/bcsj.64.2730
Return to citation in text: [1] -
Bhanu Prasad, A. S.; Bhaskar Kanth, J. V.; Periasamy, M. Tetrahedron 1992, 48, 4623–4628. doi:10.1016/S0040-4020(01)81236-9
Return to citation in text: [1] -
Das, D.; Roy, S.; Das, P. K. Org. Lett. 2004, 6, 4133–4136. doi:10.1021/ol0481176
Return to citation in text: [1] -
Barnett, J. E. G.; Kent, P. W. J. Chem. Soc. 1963, 2743–2747. doi:10.1039/JR9630002743
Return to citation in text: [1] [2] [3] [4] -
Dalla, V.; Cotelle, P.; Catteau, J. P. Tetrahedron Lett. 1997, 38, 1577–1580. doi:10.1016/S0040-4039(97)00154-8
Return to citation in text: [1] -
Dalla, V.; Catteau, J. P.; Pale, P. Tetrahedron Lett. 1999, 40, 5193–5196. doi:10.1016/S0040-4039(99)01006-0
Return to citation in text: [1] -
Soai, K.; Oyamada, H. Synthesis 1984, 605–607. doi:10.1055/s-1984-30911
Return to citation in text: [1] -
Taniguchi, M.; Fujii, H.; Oshima, K.; Utimoto, K. Tetrahedron 1993, 49, 11169–11182. doi:10.1016/S0040-4020(01)81804-4
Return to citation in text: [1] -
Padhi, S. K.; Chadha, A. Synlett 2003, 639–642. doi:10.1055/s-2003-38366
Return to citation in text: [1] [2] [3] -
Nozaki, H.; Kondô, K.; Nakanisi, O.; Sisido, K. Tetrahedron 1963, 19, 1617–1623. doi:10.1016/S0040-4020(01)99236-1
Return to citation in text: [1] [2] [3] -
Karnik, A. V.; Patil, S. T.; Patnekar, S. S.; Semwal, A. New J. Chem. 2004, 28, 1420–1422. doi:10.1039/b409061f
Return to citation in text: [1] [2] [3] [4] [5] -
Naka, T.; Koide, K. Tetrahedron Lett. 2003, 44, 443–445. doi:10.1016/S0040-4039(02)02602-3
Return to citation in text: [1] [2] [3] [4] -
Meta, C. T.; Koide, K. Org. Lett. 2004, 6, 1785–1787. doi:10.1021/ol0495366
Return to citation in text: [1] [2] [3] [4] -
Ward, R. S. Selectivity in Organic Synthesis; John Wiley and Sons: West Sussex, England, 1999; pp 4 ff.
Return to citation in text: [1] [2] [3] -
Mori, N.; Ōmura, S.; Tsuzuki, Y. Bull. Chem. Soc. Jpn. 1965, 38, 1631–1634. doi:10.1246/bcsj.38.1631
Return to citation in text: [1] [2] -
Tanner, D.; Groth, T. Tetrahedron 1997, 53, 16139–16146. doi:10.1016/S0040-4020(97)10053-9
Return to citation in text: [1] [2] -
Bhat, K. S.; Rao, A. S. Indian J. Chem. 1981, 20B, 355–358.
Return to citation in text: [1] [2] -
Mudryk, B.; Cohen, T. J. Org. Chem. 1989, 54, 5657–5659. doi:10.1021/jo00285a008
Return to citation in text: [1] -
Kamal, A.; Sandbhor, M.; Shaik, A. A. Tetrahedron: Asymmetry 2003, 14, 1575–1580. doi:10.1016/S0957-4166(03)00281-7
Return to citation in text: [1] -
Gurudutt, K. N.; Pasha, M. A.; Ravindranath, B.; Srinivas, P. Tetrahedron 1984, 40, 1629–1632. doi:10.1016/S0040-4020(01)91816-2
Return to citation in text: [1] -
Azuma, N.; Sanda, F.; Takata, T.; Endo, T. Macromolecules 1998, 31, 1710–1715. doi:10.1021/ma971082n
Return to citation in text: [1] -
Mullah, K. B.; Bentrude, W. G. J. Org. Chem. 1991, 56, 7218–7224. doi:10.1021/jo00026a008
Return to citation in text: [1] -
Shibata, T.; Fujiwara, R.; Ueno, Y. Synlett 2005, 152–154. doi:10.1055/s-2004-835664
Return to citation in text: [1] -
Kirmse, W.; Kund, K. J. Org. Chem. 1990, 55, 2325–2332. doi:10.1021/jo00295a018
Return to citation in text: [1] -
Newman, M. S. J. Org. Chem. 1961, 26, 2630–2633. doi:10.1021/jo01066a004
Return to citation in text: [1] -
Chaudhuri, S. K.; Roy, S.; Saha, M.; Bhar, S. Synth. Commun. 2007, 37, 271–274. doi:10.1080/00397910601033617
Return to citation in text: [1] -
Papageorgiou, C. D.; Ley, S. V.; Gaunt, M. J. Angew. Chem., Int. Ed. Engl. 2003, 42, 828–831. doi:10.1002/anie.200390222
Return to citation in text: [1]
| 39. | Chaudhuri, S. K.; Roy, S.; Saha, M.; Bhar, S. Synth. Commun. 2007, 37, 271–274. doi:10.1080/00397910601033617 |
| 40. | Papageorgiou, C. D.; Ley, S. V.; Gaunt, M. J. Angew. Chem., Int. Ed. Engl. 2003, 42, 828–831. doi:10.1002/anie.200390222 |
| 1. | Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, 3rd ed.; Part A and Part B; Plenum Press: New York, 1990. |
| 2. | House, H. O. Modern Synthetic Reactions; Benjamin/Cummings Publishing Company: MA, 1972. |
| 17. | Barnett, J. E. G.; Kent, P. W. J. Chem. Soc. 1963, 2743–2747. doi:10.1039/JR9630002743 |
| 22. | Padhi, S. K.; Chadha, A. Synlett 2003, 639–642. doi:10.1055/s-2003-38366 |
| 23. | Nozaki, H.; Kondô, K.; Nakanisi, O.; Sisido, K. Tetrahedron 1963, 19, 1617–1623. doi:10.1016/S0040-4020(01)99236-1 |
| 24. | Karnik, A. V.; Patil, S. T.; Patnekar, S. S.; Semwal, A. New J. Chem. 2004, 28, 1420–1422. doi:10.1039/b409061f |
| 25. | Naka, T.; Koide, K. Tetrahedron Lett. 2003, 44, 443–445. doi:10.1016/S0040-4039(02)02602-3 |
| 26. | Meta, C. T.; Koide, K. Org. Lett. 2004, 6, 1785–1787. doi:10.1021/ol0495366 |
| 27. | Ward, R. S. Selectivity in Organic Synthesis; John Wiley and Sons: West Sussex, England, 1999; pp 4 ff. |
| 1. | Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, 3rd ed.; Part A and Part B; Plenum Press: New York, 1990. |
| 2. | House, H. O. Modern Synthetic Reactions; Benjamin/Cummings Publishing Company: MA, 1972. |
| 3. | Kim, J.; Bruning, J.; Park, K. E.; Lee, D. J.; Singaram, B. Org. Lett. 2009, 11, 4358–4361. doi:10.1021/ol901677b |
| 4. | Alinezhad, H.; Tajbakhsh, M.; Zare, M. Synth. Commun. 2009, 39, 2907–2916. doi:10.1080/00397910802691882 |
| 5. | Sawada, T.; Ishii, H.; Ueda, T.; Aoki, J. Synth. Commun. 2009, 39, 3912–3923. doi:10.1080/00397910902883546 |
| 6. | Alinezhad, H.; Tajbakhsh, M.; Mahdavi, N. Synth. Commun. 2010, 40, 951–956. doi:10.1080/00397910903026731 |
| 7. | Cook, C.; Guinchard, X.; Liron, F.; Roulland, E. Org. Lett. 2010, 12, 744–747. doi:10.1021/ol902829e |
| 8. | Watanabe, T.; Imaizumi, T.; Chinen, T.; Nagumo, Y.; Shibuya, M.; Usui, T.; Kanoh, N.; Iwabuchi, Y. Org. Lett. 2010, 12, 1040–1043. doi:10.1021/ol1000389 |
| 9. | McIver, A. L.; Deiters, A. Org. Lett. 2010, 12, 1288–1291. doi:10.1021/ol100177u |
| 10. | Collins, J.; Rinner, U.; Moser, M.; Hudlicky, T.; Ghiviriga, I.; Romero, A. E.; Kornienko, A.; Ma, D.; Griffin, C.; Pandey, S. J. Org. Chem. 2010, 75, 3069–3084. doi:10.1021/jo1003136 |
| 16. | Das, D.; Roy, S.; Das, P. K. Org. Lett. 2004, 6, 4133–4136. doi:10.1021/ol0481176 |
| 14. | Yamakawa, T.; Masaki, M.; Nohira, H. Bull. Chem. Soc. Jpn. 1991, 64, 2730–2734. doi:10.1246/bcsj.64.2730 |
| 15. | Bhanu Prasad, A. S.; Bhaskar Kanth, J. V.; Periasamy, M. Tetrahedron 1992, 48, 4623–4628. doi:10.1016/S0040-4020(01)81236-9 |
| 31. | Mudryk, B.; Cohen, T. J. Org. Chem. 1989, 54, 5657–5659. doi:10.1021/jo00285a008 |
| 32. | Kamal, A.; Sandbhor, M.; Shaik, A. A. Tetrahedron: Asymmetry 2003, 14, 1575–1580. doi:10.1016/S0957-4166(03)00281-7 |
| 13. | Soai, K.; Oyamada, H.; Ookawa, A. Synth. Commun. 1982, 12, 463–467. doi:10.1080/00397918208065953 |
| 1. | Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, 3rd ed.; Part A and Part B; Plenum Press: New York, 1990. |
| 2. | House, H. O. Modern Synthetic Reactions; Benjamin/Cummings Publishing Company: MA, 1972. |
| 17. | Barnett, J. E. G.; Kent, P. W. J. Chem. Soc. 1963, 2743–2747. doi:10.1039/JR9630002743 |
| 22. | Padhi, S. K.; Chadha, A. Synlett 2003, 639–642. doi:10.1055/s-2003-38366 |
| 23. | Nozaki, H.; Kondô, K.; Nakanisi, O.; Sisido, K. Tetrahedron 1963, 19, 1617–1623. doi:10.1016/S0040-4020(01)99236-1 |
| 24. | Karnik, A. V.; Patil, S. T.; Patnekar, S. S.; Semwal, A. New J. Chem. 2004, 28, 1420–1422. doi:10.1039/b409061f |
| 25. | Naka, T.; Koide, K. Tetrahedron Lett. 2003, 44, 443–445. doi:10.1016/S0040-4039(02)02602-3 |
| 26. | Meta, C. T.; Koide, K. Org. Lett. 2004, 6, 1785–1787. doi:10.1021/ol0495366 |
| 27. | Ward, R. S. Selectivity in Organic Synthesis; John Wiley and Sons: West Sussex, England, 1999; pp 4 ff. |
| 11. | Brown, H. C.; Mead, E. J.; Subba Rao, B. C. J. Am. Chem. Soc. 1955, 77, 6209–6213. doi:10.1021/ja01628a044 |
| 12. | Brown, M. S.; Rapoport, H. J. Org. Chem. 1963, 28, 3261–3263. doi:10.1021/jo01046a538 |
| 28. | Mori, N.; Ōmura, S.; Tsuzuki, Y. Bull. Chem. Soc. Jpn. 1965, 38, 1631–1634. doi:10.1246/bcsj.38.1631 |
| 29. | Tanner, D.; Groth, T. Tetrahedron 1997, 53, 16139–16146. doi:10.1016/S0040-4020(97)10053-9 |
| 21. | Taniguchi, M.; Fujii, H.; Oshima, K.; Utimoto, K. Tetrahedron 1993, 49, 11169–11182. doi:10.1016/S0040-4020(01)81804-4 |
| 1. | Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, 3rd ed.; Part A and Part B; Plenum Press: New York, 1990. |
| 2. | House, H. O. Modern Synthetic Reactions; Benjamin/Cummings Publishing Company: MA, 1972. |
| 17. | Barnett, J. E. G.; Kent, P. W. J. Chem. Soc. 1963, 2743–2747. doi:10.1039/JR9630002743 |
| 23. | Nozaki, H.; Kondô, K.; Nakanisi, O.; Sisido, K. Tetrahedron 1963, 19, 1617–1623. doi:10.1016/S0040-4020(01)99236-1 |
| 24. | Karnik, A. V.; Patil, S. T.; Patnekar, S. S.; Semwal, A. New J. Chem. 2004, 28, 1420–1422. doi:10.1039/b409061f |
| 25. | Naka, T.; Koide, K. Tetrahedron Lett. 2003, 44, 443–445. doi:10.1016/S0040-4039(02)02602-3 |
| 26. | Meta, C. T.; Koide, K. Org. Lett. 2004, 6, 1785–1787. doi:10.1021/ol0495366 |
| 27. | Ward, R. S. Selectivity in Organic Synthesis; John Wiley and Sons: West Sussex, England, 1999; pp 4 ff. |
| 24. | Karnik, A. V.; Patil, S. T.; Patnekar, S. S.; Semwal, A. New J. Chem. 2004, 28, 1420–1422. doi:10.1039/b409061f |
| 18. | Dalla, V.; Cotelle, P.; Catteau, J. P. Tetrahedron Lett. 1997, 38, 1577–1580. doi:10.1016/S0040-4039(97)00154-8 |
| 19. | Dalla, V.; Catteau, J. P.; Pale, P. Tetrahedron Lett. 1999, 40, 5193–5196. doi:10.1016/S0040-4039(99)01006-0 |
| 17. | Barnett, J. E. G.; Kent, P. W. J. Chem. Soc. 1963, 2743–2747. doi:10.1039/JR9630002743 |
| 33. | Gurudutt, K. N.; Pasha, M. A.; Ravindranath, B.; Srinivas, P. Tetrahedron 1984, 40, 1629–1632. doi:10.1016/S0040-4020(01)91816-2 |
| 28. | Mori, N.; Ōmura, S.; Tsuzuki, Y. Bull. Chem. Soc. Jpn. 1965, 38, 1631–1634. doi:10.1246/bcsj.38.1631 |
| 29. | Tanner, D.; Groth, T. Tetrahedron 1997, 53, 16139–16146. doi:10.1016/S0040-4020(97)10053-9 |
| 25. | Naka, T.; Koide, K. Tetrahedron Lett. 2003, 44, 443–445. doi:10.1016/S0040-4039(02)02602-3 |
| 24. | Karnik, A. V.; Patil, S. T.; Patnekar, S. S.; Semwal, A. New J. Chem. 2004, 28, 1420–1422. doi:10.1039/b409061f |
| 36. | Shibata, T.; Fujiwara, R.; Ueno, Y. Synlett 2005, 152–154. doi:10.1055/s-2004-835664 |
| 37. | Kirmse, W.; Kund, K. J. Org. Chem. 1990, 55, 2325–2332. doi:10.1021/jo00295a018 |
| 34. | Azuma, N.; Sanda, F.; Takata, T.; Endo, T. Macromolecules 1998, 31, 1710–1715. doi:10.1021/ma971082n |
| 35. | Mullah, K. B.; Bentrude, W. G. J. Org. Chem. 1991, 56, 7218–7224. doi:10.1021/jo00026a008 |
© 2010 Chaudhuri et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)