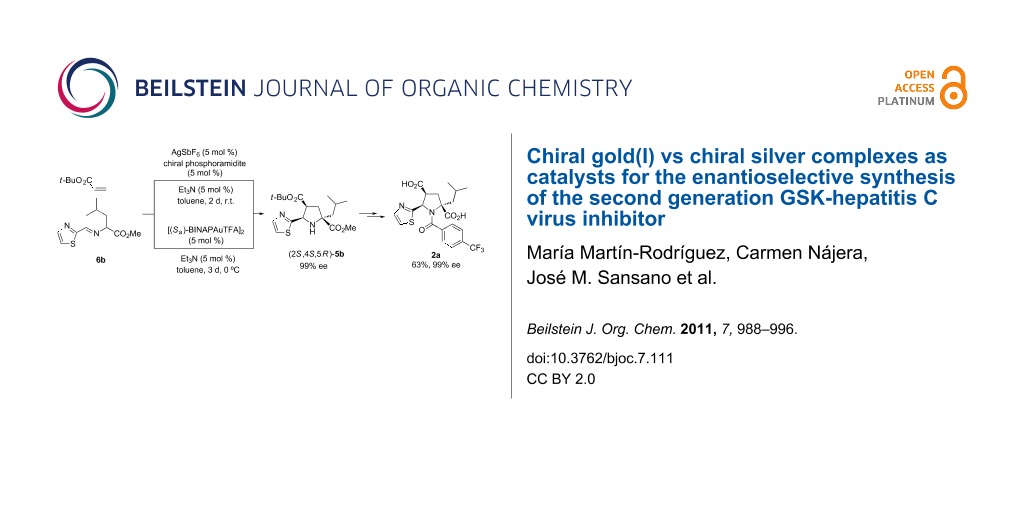
Abstract
The synthesis of a GSK 2nd generation inhibitor of the hepatitis C virus, by enantioselective 1,3-dipolar cycloaddition between a leucine derived iminoester and tert-butyl acrylate, was studied. The comparison between silver(I) and gold(I) catalysts in this reaction was established by working with chiral phosphoramidites or with chiral BINAP. The best reaction conditions were used for the total synthesis of the hepatitis C virus inhibitor by a four step procedure affording this product in 99% ee and in 63% overall yield. The origin of the enantioselectivity of the chiral gold(I) catalyst was justified according to DFT calculations, the stabilizing coulombic interaction between the nitrogen atom of the thiazole moiety and one of the gold atoms being crucial.

Graphical Abstract
Introduction
The prevalence of chronic hepatitis C virus (HCV) infection is such that it is estimated to be suffered by around 200 million people worldwide [1]. This enveloped single-stranded RNA virus (belonging to the Flaviviridae family) is present in six major genotypes in the world’s industrialized nations, genotype 1 being the most prevalent, followed by genotype 2 and 3. Due to the poor toleration of the current therapy, and the lack of an appropriate vaccine, researchers working on strategies for developing antivirals have tried to attack viruses at every stage of their life cycles, namely attachment to a host cell, replication of viral components, assembly of viral components into complete viral particles and release of viral particles able to infect new hosts cells. Inside the infected hepatocytes, structural E1 and E2 and non-structural proteins such as NS2, NS3 (which bear serine proteinase, helicase, and NTPase activities), NS4A, NS4B, NS5A (regulators of RNA replication), and NS5B (the RNA-dependent RNA polymerase) are generated [2,3] and, in fact, constitute the main targets. At the moment, there are many drugs under clinical trial evaluation, the compounds targeting HCV replication being the most promising candidates to achieve a sustained virological response [1,4]. Several years ago, a high-throughput screening of the GlaxoSmithKline compound collection identified a series of small pyrrolidine molecules, e.g., 1 (Figure 1), able to inhibit the RNA-dependent RNA polymerase of the virus responsible for hepatitis C (genotype 1g) [5]. Thus, their high replication rates (billions of copies per day) can be drastically suppressed by the inhibition of the NS5B RNA-dependent RNA polymerase enzyme, which is the primary target for oral antiviral agents [6,7]. In further studies, a second generation of antiviral agents 2 and 3 (Figure 1), offering a greater dynamic range even for HCV genotype 1b, was published [5,8,9]. These molecules incorporated a 2-thiazole heterocycle instead of the 2-thienyl group, together with a more hydrophobic environment at the amido group [9-12]. However, the design of improved broader spectrum compounds, capable of effective inhibition of genotypes 1a and 1b, is desirable. In this sense, GSK625433 (4) (Figure 1) has exhibited a good pharmacokinetic profile in preclinical animal species [13].
![[1860-5397-7-111-1]](/bjoc/content/figures/1860-5397-7-111-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: More active GSK HCV inhibitors.
Figure 1: More active GSK HCV inhibitors.
The synthesis of the endo-pyrrolidine core of 5 is the key step for the preparation of these antiviral agents, and can be efficiently achieved by a 1,3-dipolar cycloaddition (1,3-DC) between the corresponding azomethine ylide and an alkyl acrylate [14-18] (Scheme 1). The first synthesis of racemic product 1, and other derivatives including compounds 2, was achieved in several steps using, as the key reaction, the silver(I) or lithium(I)-metalloazomethine ylide, under basic conditions, and tert-butyl acrylate. The enantiomeric samples were isolated by semi-preparative chiral HPLC [9,10]. The first endo-diastereoselective synthesis of the key precursor 5a (HetAr = 2-thienyl), of the antiviral agent 1 (96% de), was achieved by our group from imine 6a (HetAr = 2-thienyl; R1 = Me) in the presence of the acrylate derived from (R)-methyl lactate [19]. However, the most straightforward, and also faster, approach to the enantiomeric formation of this non-nucleosidic antiviral agent 1 is based on a catalytic enantioselective 1,3-DC [20-24]. The first reported enantioselective overall synthesis of the structure 1 was catalyzed by a chiral phosphoramidite and AgClO4 [25,26], although the synthesis of the five-membered core has also been published using chiral calcium complexes [27,28].
![[1860-5397-7-111-i1]](/bjoc/content/inline/1860-5397-7-111-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of antiviral structures.
Scheme 1: Retrosynthetic analysis of antiviral structures.
In addition, for the second generation antivirals 2 or 3, the efficiency of the Lewis acid-catalyzed 1,3-DC, following the route shown in Scheme 1, was combined with hydroquinine as chiral base (6 mol %) together with silver acetate (3 mol %), and this afforded moderate enantioselectivities (70–74%) of 5b, in such a way that a further 1,1’-binaphthyl-2,2’-dihydrogen phosphate assisted chiral resolution was required to increase the optical purity of the target molecule [11]. Chiral calcium(II) complexes have been used for the synthesis of a similar key molecule 5b (R1 = t-Bu, 88% ee), but the overall synthesis of the antiviral drug was not reported [27,28].
In this article, we describe the full study concerning the enantioselective synthesis of product 5b using silver(I) or gold(I) complexes, generated from chiral phosphoramidites or BINAP as ligands, in order to prepare antiviral agent 2a.
Results and Discussion
The efficiency of the chiral phosphoramidite/silver(I) salts [25,26,29] and BINAP/Ag(I) salts [30,31] in 1,3-DC, following the general pattern shown in Scheme 1, has been demonstrated by our group, establishing a wider scope and sensibly higher enantioselectivities for the reactions performed in the presence of chiral phosphoramidite/silver(I) complexes [24]. Concerning enantioselective gold(I)-catalyzed 1,3-DC, the classical cycloaddition starting from iminoesters 6 has not been so extensively explored. Reports of chiral transformations involving azlactones [32,33] and iminoesters 6 [34], which employed chiral diphosphines and gold(I) salts, have been published showing very good endo-diastereoselectivities and moderate to excellent enantioselectivities. However, the use of acrylates as dipolarophiles has only been explored with the 2-thienyliminoesters 6a.
Therefore, based on our experience of silver(I)- and gold(I)-catalyzed 1,3-DC involving azomethine ylides derived from α-iminoester 6b and tert-butyl acrylate, we selected a series of known chiral phosphoramidite ligands (Figure 2), which were prepared according to the literature [35]. The chiral phosphoramidite/silver(I) complexes were generated in situ by mixing equimolar amounts of both components at room temperature for 30 min. Chiral phosphoramidite/AuCl complexes were generated according to the literature [36] and, finally, underwent anion interchange in the presence of the corresponding silver salt. The precipitate was filtered through a celite pad and used without any other additional treatment.
![[1860-5397-7-111-2]](/bjoc/content/figures/1860-5397-7-111-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Chiral phosphoramidites tested in this study.
Figure 2: Chiral phosphoramidites tested in this study.
All of the reactions were performed at room temperature, employing a 5 mol % of both catalyst and base, for 17 h (Scheme 1). Reactions between iminoester 6b and tert-butyl acrylate, which employed silver complexes derived from Monophos (Sa)-7 ligand, afforded racemic endo-cycloadduct 5b (Table 1, entries 1–3). The analogous reaction catalyzed by chiral phosphoramidite 7/gold(I) complexes did not occur at all when AgClO4 or AgSbF6 were employed as anion interchange agents. Just a small conversion, with some side products, and null enantioselectivity was observed in the crude reaction mixtures obtained when using (Sa)-7/AuTFA (Table 1, entry 4). When the reaction was carried out in the presence of chiral ligand (Ra,R)-8 the enantioselectivities were low or moderate in the examples concerning AgClO4 and AgTFA (TFA = trifluoroacetate anion), respectively (Table 1, entries 5 and 7). Surprisingly, the reaction involving this chiral ligand 8 combined with AgSbF6 afforded a good yield of the enantiomerically pure cycloadduct 5b (Table 1, entry 6). Attempts to increase the enantioselectivity, in the example run with AgTFA, by replacing triethylamine by diisopropylethylamine (DIPEA) were not successful, and only a slight increment of enantiomeric excess was observed (Table 1, entry 8). Again, the gold complex (Ra,R)-8/AuTFA did not give the expected reaction product (Table 1, entry 9). The employment of this matched combination with (Ra,R)-8 was justified by the low enantioselectivity achieved through the use of (Ra,S)-8 in the same transformation (not shown in Table 1). The widely used chiral ligand (Sa,R,R)-9 has also been similarly studied. In this case, the matched combination was determined in previous works that investigated the scope of enantioselective silver(I)-catalyzed 1,3-DC of azomethine ylides and dipolarophiles [25,29]. The enantioselectivities were moderate, even when using AgSbF6, and the effect of the added base was negligible (Table 1, entries 10–14). The process catalyzed by the (Sa,R,R)-9/AuTFA was not suitable (Table 1, entry 15). The more sterically hindered chiral phosphoramidite (Sa,R,R)-10 did not afford any interesting results because the conversions were extremely low after 2 days reaction, and the crude reaction mixture was very complex (1H NMR analysis) (Table 1, entries 16–18). However, a good result was obtained when phosphoramidite (Sa,R,R)-11 was tested together with AgClO4. The high enantioselectivity achieved for 5b (86% ee) is in contrast to the racemic samples identified when either AgSbF6 or AgTFA were employed as co-catalysts (Table 1, entries 19–21). Biphenol derived ligand (R,R)-12 generally furnished good yields of the cycloadduct 5b but with a low enantiodiscrimination (Table 1, entries 22–24). In many examples, although the reactions were performed at lower temperatures (0 or −20 °C, not shown in Table 1) the resulting enantioselectivities did not suffer noticeable variations. In all of the cases given in Table 1, the endo-cycloadduct was exclusively generated, and the absolute configuration of 5b was established by extrapolation with the results previously obtained for each chiral catalyst [25,26,28-30,33]. According to these results the combination of chiral phosphoramidite and silver(I) salt is much more appropriate than the analogous one made with gold(I) salts. Especially useful is the reaction of (Ra,R)-8/AgSbF6 catalytic complex affording enantiomerically pure cycloadduct endo-5b. It is worth mentioning that chiral phosphoramidite/gold(I) complexes, formed by anion interchange of the corresponding phosphoramidite/AuCl complex and AgSbF6 [36] or AgBF4 [37,38], have been successfully employed in enantioselective cycloaddition of allenedienes [36,37] or allenenes [38] under very mild reaction conditions (0 ºC to r.t.). Despite these described opportunities provided by chiral phosphoramidite ligands as a part of gold(I) complexes, their activity (see Table 1) was negligible, until now, when applied in the 1,3-DC represented in Scheme 2.
Table 1: Optimization of the 1,3-dipolar cycloaddition of 6b and tert-butyl acrylate using chiral phosphoramidite ligands.
| Entry | Catalysta | Base | Yieldb (%) | eec (%) |
|---|---|---|---|---|
| 1 | (Sa)-7/AgClO4 | Et3N | ___d | rac |
| 2 | (Sa)-7/AgSbF6 | Et3N | ___d | rac |
| 3 | (Sa)-7/AgTFA | Et3N | ___d | rac |
| 4 | (Sa)-7/AuTFA | DIPEA | ___d | ___d |
| 5 | (Sa,R)-8/AgClO4 | Et3N | 82 | 20 |
| 6 | (Sa,R)-8/AgSbF6 | Et3N | 82 | 99 |
| 7 | (Sa,R)-8/AgTFA | Et3N | 82 | 60 |
| 8 | (Sa,R)-8/AgTFA | DIPEA | 82 | 64 |
| 9 | (Sa,R)-8/AuTFA | DIPEA | ___d | ___d |
| 10 | (Sa,R,R)-9/AgClO4 | DIPEA | 86 | 30 |
| 11 | (Sa,R,R)-9/AgSbF6 | Et3N | 72 | 40 |
| 12 | (Sa,R,R)-9/AgSbF6 | DIPEA | 82 | 40 |
| 13 | (Sa,R,R)-9/AgTFA | Et3N | 82 | 50 |
| 14 | (Sa,R,R)-9/AgTFA | DIPEA | 82 | 40 |
| 15 | (Sa,R,R)-9/AuTFA | DIPEA | ___d | ___d |
| 16 | (Sa,R,R)-10/AgClO4 | Et3N | ___d | rac |
| 17 | (Sa,R,R)-10/AgTFA | Et3N | ___d | rac |
| 18 | (Sa,R,R)-10/AgSbF6 | Et3N | ___d | rac |
| 19 | (Sa,R,R)-11/AgClO4 | Et3N | 72 | 86 |
| 20 | (Sa,R,R)-11/AgSbF6 | Et3N | ___d | rac |
| 21 | (Sa,R,R)-11/AgTFA | Et3N | ___d | rac |
| 22 | (R,R)-12/AgClO4 | Et3N | 79 | 30 |
| 23 | (R,R)-12/AgTFA | Et3N | 87 | 40 |
| 24 | (R,R)-12/AgSbF6 | Et3N | 86 | 30 |
aThe generation of silver catalysts was achieved by mixing equimolar amounts of silver(I) or gold(I) salt and the corresponding phosphoramidite. bAfter flash chromatography (silica gel). The observed endo:exo ratio was always >98:2 (1H NMR). cDetermined by using analytical chiral HPLC columns (Daicel, Chiralpak AS). dNot determined.
![[1860-5397-7-111-i2]](/bjoc/content/inline/1860-5397-7-111-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Optimization of the reaction conditions for the synthesis of the key intermediate 5b.
Scheme 2: Optimization of the reaction conditions for the synthesis of the key intermediate 5b.
The chiral ligand (Sa)-BINAP (13) was also tested in the standard reaction to access key molecule endo-5b (Scheme 3). AgClO4 was found to be the most appropriate silver salt to achieve the highest enantioselectivity (88% ee) compared to the results obtained when other silver salts were employed (Table 2, entries 1, 3, and 4). In agreement with the previous results, the reaction with chiral silver complexes at lower temperatures did not improve the enantioselectivity. According to our previous work, dimeric chiral gold(I) catalyst [(Sa)-BINAPAuTFA]2 (Sa,Sa)-14 was very efficient in 1,3-DC compared to other catalysts with different stoichiometry or anion nature. The gold complex (Sa,Sa)-14 was prepared according to the literature [39] and immediately used in the cycloaddition in the absence of base because of its bifunctional behaviour, namely the activation of the basic character of the dipole [34]. However, no reaction occurred under these conditions (Table 2, entry 5). Therefore, the presence of the base was crucial for the evolution of the reaction, as can be seen in entries 6 and 7 of Table 2. Triethylamine promoted the reaction affording good yield and good enantioselectivity (78% ee). However, DIPEA-mediated cycloaddition did not improve the enantioselectivity of the resulting endo-cycloadduct 5b. Unlike the results obtained with silver(I) catalytic complexes at lower temperatures (0 or −20 °C), the gold(I)-catalyzed cycloaddition could be successfully carried out at 0 °C resulting in excellent enantiodiscrimination (99% ee) to the detriment of the reaction time, which had to be increased to 3 days (Table 2, entry 8). The result obtained in this last example was excellent but the enantiomeric excess achieved at room temperature in the reaction performed with (Sa)-13/AgClO4 complex is also valuable.
![[1860-5397-7-111-i3]](/bjoc/content/inline/1860-5397-7-111-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of the enantiomerically enriched 5b.
Scheme 3: Preparation of the enantiomerically enriched 5b.
Table 2: Optimization of the 1,3-dipolar cycloaddition of 6a and tert-butyl acrylate using chiral (Sa)-BINAP (13) ligand.
| Entry | Catalysta | Base | Yieldb (%) | eec (%) |
|---|---|---|---|---|
| 1 | (Sa)-13/AgClO4 | Et3N | 78 | 88 |
| 2 | (Sa)-13/AgClO4 | Et3Nd | 75 | 85 |
| 3 | (Sa)-13/AgSbF6 | Et3N | 79 | 72 |
| 4 | (Sa)-13/AgTFA | Et3N | 82 | 40 |
| 5 | (Sa,Sa)-14 | ___ | ___e | ___e |
| 6 | (Sa,Sa)-14 | Et3N | 90 | 78 |
| 7 | (Sa,Sa)-14 | DIPEA | 87 | 70 |
| 8 | (Sa,Sa)-14 | Et3Nd,f | 92 | 99 |
aThe generation of silver catalysts was achieved by mixing equimolar amounts of silver(I) and (Sa)-BINAP. bAfter flash chromatography (silica gel). The observed endo:exo ratio was always >98:2 (1H NMR). cDetermined using analytical chiral HPLC columns (Daicel, Chiralpak AS). dReaction performed at 0 °C. eNot determined. fAfter 3 days reaction.
With the most enantiomerically enriched cycloadduct 5b, the synthesis of the antiviral agent 2a could be accomplished in two conventional steps involving an amidation reaction and a double ester hydrolysis. The latter step consisted of a first stage TFA-mediated hydrolysis of the tert-butyl ester followed by a basic stage employing a refluxing solution of KOH/MeOH (Scheme 4). The final product 2b was finally isolated in 68% overall yield (from pyrrolidine 5b) and with 99% ee, or alternatively in 63% overall yield from iminoester 6b.
![[1860-5397-7-111-i4]](/bjoc/content/inline/1860-5397-7-111-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Total synthesis of antiviral agent 2b.
Scheme 4: Total synthesis of antiviral agent 2b.
Although the study of the enantioselectivity exhibited by chiral phosphoramidite/silver(I) complexes employing DFT calculations was confirmed by our group [25], an explanation for the excellent results obtained employing the gold complex (Sa,Sa)-14 (Table 2, entry 8) was needed. In a previous work, we demonstrated that the stereoselectivity of the 1,3-DC employing chiral metallic Lewis bases arises from the blockage of one of the prochiral faces [40]. In this way, our results (in terms of DFT calculations) show that there is only one energetically accessible conformation due to the high substitution of the leucine-derived ylide (Figure 3). In this reactive complex there is an effective blockage of the (2re,5si) prochiral face of the ylide. Therefore, the predicted stereochemical outcome corresponds to the exclusive formation of the (2S,4S,5R)-5b cycloadduct, the same as that obtained experimentally.
![[1860-5397-7-111-3]](/bjoc/content/figures/1860-5397-7-111-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Gibbs activation energy and main geometrical features of the computed ylide and transition structures (TS) corresponding to the 1,3-DC of the Au(I)–ylide complex and tert-butyl acrylate computed at ONIOM(B3LYP/LanL2DZ:UFF) level of theory. High-level and low-level layers are represented as ball & stick and wireframe models, respectively. Grey numbers in parentheses represent Mulliken charges. Distances are in Å.
Figure 3: Gibbs activation energy and main geometrical features of the computed ylide and transition structur...
As shown in Figure 3, the reaction proceeds to a concerted but highly asynchronous cycloaddition in which the endo-approach of the dipolarophile is favoured due to a stabilizing interaction of the carboxylic group and the metallic centre. The computed activation Gibbs free energy barrier associated with the formation of (2S,4S,5R)-5b is 23.4 kcal mol−1, which means that the process is feasible at the reaction temperature. It is worth noting that there is a stabilizing coulombic interaction between the nitrogen atom of the thiazole moiety (N7) and one of the gold atoms of the catalyst, both in the TS and the ylide complex. This interaction fixes the planar conformation of the ylide moiety and minimizes the possible steric hindrance with the bulky tert-butyl group of the dipolarophile. When a phenyl substituent is placed to the imino group this planar conformation does not exist and, in consequence, a more steric interaction avoids the approach of the mentioned dipolarophile.
Conclusion
In this work the complexity of the 1,3-DC reaction of azomethine ylides and dipolarophiles (in this case acrylates) was demonstrated. There are many parameters to control and a small variation can cause a dramatic effect in the overall enantiodiscrimination of the process. The temperature does not equally affect silver(I) and gold(I) catalysts. The effect of the heterocycle remains crucial in these transformations because, originally, the enantioselectivity of the reaction between methyl benzylideneiminoglycinate and alkyl acrylates failed in the presence of the silver(I) or the dimeric gold(I) complexes derived from chiral BINAP. The metal cation and the counterion are also important in the final result and, in certain cases, their position with respect to the reaction centre can modify the overall reaction and consequently alter the enantioselectivity of the process. To date, the best reaction conditions to access GSK 2nd generation antiviral drugs 2a are: The employment of chiral phosphoramidite (Ra,R)-8/AgSbF6 and Et3N (both in 5 mol % amount) at r.t. for 2 h, or chiral (Sa,Sa)-14 gold complex and Et3N (both in 5 mol % amount) at 0 °C for 3 days. Whilst phosphoramidite complexes operated exclusively in the presence of silver salts, the most versatile chiral BINAP ligand could work efficiently with both silver(I) or gold(I) cations. The stabilizing coulombic interaction between the nitrogen atom of the thiazole moiety and one of the gold atoms of the catalyst both in the TS and the ylide complex is the explanation for the success of the gold-catalyzed cycloaddition, in constrast to the observed TS involving methyl benzylideneiminoleucinate.
Experimental
General. All reactions were carried out in the absence of light. Anhydrous solvents were freshly distilled under an argon atmosphere. Aldehydes were also distilled prior to use for the elaboration of the iminoesters. Melting points were determined with a Reichert Thermovar hot plate apparatus and are uncorrected. Only the structurally most important peaks of the IR spectra (recorded on a Nicolet 510 P-FT and on a Jasco FTIR 4100) are listed. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were obtained on a Bruker AC-300 using CDCl3 as solvent and TMS as internal standard, unless otherwise stated. Optical rotations were measured on a Perkin Elmer 341 polarimeter. HPLC analyses were performed on a JASCO 2000-series equipped with a chiral column (detailed for each compound in the main text), using mixtures of n-hexane/isopropyl alcohol as mobile phase, at 25 °C. Low-resolution electron impact (EI) mass spectra were obtained at 70 eV on a Shimadzu QP-5000 and high-resolution mass spectra were obtained on a Finnigan VG Platform. HRMS (EI) were recorded on a Finnigan MAT 95S. Microanalyses were performed on a Perkin Elmer 2400 and a Carlo Erba EA1108. Analytical TLC was performed on Schleicher & Schuell F1400/LS silica gel plates and the spots were visualized under UV light (λ = 254 nm). For flash chromatography we employed Merck silica gel 60 (0.040–0.063 mm). Ligands 7–12 were prepared according to the reported procedure (see text). All of the transformations performed with silver catalysts were performed in the absence of light. The synthesis of the already characterized chiral complex (Sa,Sa)-14 was performed according to the published procedure [39].
Computational methods. Hybrid QM/MM calculations for optimizations of saddle points were performed in terms of ONIOM [41-43] method implemented in GAUSSIAN09 suite of programs [44]. Ball & stick model in Figure 3 shows atoms included in the high-level layer, and a wire model is used to represent atoms included in the low-level layer. In the high-level layer, the electron correlation was partially taken into account by using the hybrid functional B3LYP [45-50] combined with Hay-Wadt small core effective potential (ECP) [51] basis set. UFF [52] molecular mechanics force field was employed in the low-level layer. Thermal corrections of Gibbs free energies were computed at the same level of theory and were not scaled. All stationary points were characterized by harmonic analysis. Reactant intermediates and cycloadducts have positive definite Hessian matrices. Transition structures show only one negative eigenvalue in their diagonalized force constant matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration.
1,3-Dipolar cycloaddition of iminoester 6b and tert-butyl acrylate. General procedure. To a solution of the in situ prepared chiral gold complex or chiral silver complex (0.05 mmol) in toluene (2 mL) was added, at r.t., a solution of the iminoester 6b (120 mg, 0.5 mmol) and tert-butyl acrylate (109 μL, 0.75 mmol) in toluene (2 mL). In some cases DIPEA or triethylamine (0.05 mmol) was added (see Tables) and the mixture stirred at r.t. or 0 °C for 2 or 3 days (see Tables). The reaction mixture was filtered off through a celite pad, the organic filtrate was directly evaporated and the residue was purified by recrystallization or by flash chromatography, yielding pure endo-cycloadduct 5b.
(2S,4S,5R)-4-tert-Butyl-2-methyl-2-isobutyl-5-(thiazol-2-yl)pyrrolidine-2,4-dicarboxylate (5b): Colourless solid; mp >195 °C dec (n-hexane/ethyl acetate); [α]D20 +43 (c 1.00, CH2Cl2, 99% ee by HPLC); IR (neat) νmax: 3330, 1718 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.70, 7.27 (2 × d, J = 3.4 Hz, 2H, CHCHS), 4.83 (d, J = 7.5 Hz, 1H, CHCS), 3.73 (s, 3H, OCH3), 3.41 (q, J = 7.8 Hz, 1H, CHCHN), 3.25 (br. s, 1H, NH), 2.79, 2.10 (2 × dd, J = 13.5, 7.9 Hz, 2H, CH2CCO), 1.76–1.69 (m, 2H, CH2CH), 1.54–1.48 (m, 1H, CH2CH), 1.17 (s, 9H, (CH3)3), 0.94, 0.85 (2 × d, J = 6.2 Hz, 6H, 2 × CH3C); 13C NMR (75 MHz, CDCl3) δ 176.2, 170.8, 170.6 (2 × CO and CSN), 142.4, 118.8 (CHCHS), 80.6 (C(CH3)3), 68.3 (COCN), 61.7 (CHCS), 52.2 (OCH3), 49.6 (CHCO), 49.3 (CH2CCO), 39.5 (CH2CH), 27.6 ((CH3)3), 25.0 (C(CH3)2), 24.3, 22.9 (2 × CH3C); EIMS m/z (% relative intensity): 368 (M+, 1), 310 (51), 295 (16), 255 (23), 254 (14), 253 (100); HRMS calcd for C18H28N2O4S, 368.1770; found, 368.1761; HPLC (Chiralpak AD-H), n-hexane:iPrOH 95/5, 1 mL/min, λ = 225 nm, tR,maj = 12 min, tR,min = 18 min.
Synthesis of the antiviral agent 2b. Compound (2S,4S,5R)-5b (1.2 mmol, 441 mg) was dissolved in dichloromethane (25 mL), and pyridine (2.4 mmol, 174 μL) and 4-(trifluoromethyl)benzoyl chloride (1.2 mmol, 182 μL) were slowly added at 0 °C. The resulting mixture was refluxed for 1 day and the solvent was removed under vacuo (15 Torr). Crude compound (2S,4S,5R)-15, was allowed to react with trifluoroacetic acid/dichloromethane mixture (9.6 mL/18 mL). The resulting mixture was stirred at r.t. overnight and the solvent evaporated under vacuo. The residue was dissolved in a 1 M solution of KOH in a 4/1 MeOH/H2O (50 mL) and refluxed for 16 h. Methanol was evaporated and aqueous HCl (0.5 M, 20 mL) and ethyl acetate were added (2 × 20 mL). The combined organic phases were dried (MgSO4) and evaporated, yielding the crude compound (2S,4S,5R)-2b, which was recrystallized from a mixture containing n-hexane/ethyl acetate.
(2S,4S,5R)-2-Isobutyl-5-(thiazol-2-yl)-1-[4-(trifluoromethyl)benzoyl]pyrrolidine-2,4-dicarboxylic acid (2b): Pale brown solid; mp >130 °C dec (n-hexane/ethyl acetate); [α]D20 +35 (c 0.3, toluene, 99% ee); IR (neat) νmax: 3100, 1731, 1693 cm−1; 1H NMR (300 MHz, CD3COCD3) δ 8.15 (d, J = 7.5 Hz, 2H, ArH), 7.80–7.64 (m, 3H, ArH and CHCHS), 7.29 (d, J = 3.4 Hz, 1H, CHCHS), 5.85 (d, J = 8.7 Hz, 1H, CHNS), 4.01–3.81 (m, 1H, CHCO), 2.84 (t, J = 13.3 Hz, 1H, CH2CCO), 2.34 (dd, J = 13.2, 6.5 Hz, 1H, CH2CCO), 1.28 (m, 4H, CH2CH and 2 × OH), 1.14–1.06 (m, 1H, CH2CH), 0.85 (m, 6H, 2 × CH3C); 13C NMR (75 MHz, CDCl3) δ 172.4, 169.1, 168.9, 167.4 (3 × CO and CNS), 141.1, 134.2, 134.1, 130.2, 126.9, 125.4, 120.9, (ArC, CF3, and CHCHS), 69.7 (COCN), 65.3 (NCH), 51.39 (CHCO), 42.3 (CH2CCO), 35.3 (CH2CH), 25.7 (CH(CH3)2), 24.4, 24.2 (CH(CH3)2); ESIMS m/z (% relative intensity) 470 (M+, 2); HRMS calcd for C21H21F3N2O5S, 470.4620; found, 470,4631; HPLC (Chiralpak AD-H), n-hexane:iPrOH 85/15, 0.1 mL/min, λ = 250 nm), tR,maj = 12.5 min, tR,min = 15.5 min.
Acknowledgements
This work has been supported by the DGES of the Spanish Ministerio de Ciencia e Innovación (MICINN) (Consolider INGENIO 2010 CSD2007-00006, FEDER-CTQ2007-62771/BQU, CTQ2007/67528, CTQ2010-20387 and by the Hispano-Brazilian project PHB2008-0037-PC), Generalitat Valenciana (PROMETEO/ 2009/039), the Basque government (Grant IT-324-07) and by the University of Alicante. M. M.-R. Also thanks DGES for a grant. The authors also thank the SGI/IZO-SGIker of UPV/EHU for allocation of computational resources.
References
-
Gao, M.; Nettles, R. E.; Belema, M.; Snyder, L. B.; Nguyen, V. N.; Fridell, R. A.; Serrano-Wu, M. H.; Langley, D. R.; Sun, J.-H.; O’Boyle, D. R., II; Lemm, J. A.; Wang, C.; Knipe, J. O.; Chien, C.; Colonno, R. J.; Grasela, D. M.; Meanwell, N. A.; Hamann, L. G. Nature 2010, 465, 96–100. doi:10.1038/nature08960
Return to citation in text: [1] [2] -
Tellinghuisen, T. L.; Evans, M. J.; von Hahn, T.; You, S.; Rice, C. M. J. Virol. 2007, 81, 8853–8867. doi:10.1128/JVI.00753-07
Return to citation in text: [1] -
Gish, R. Future Therapies for Hepatitis C. In The HCV Advocate Medical Writers’ Circle; Franciscus, A., Ed.; HCSP Publications: San Francisco, February 1, 2006.
Return to citation in text: [1] -
Lange, C. M.; Sarrazin, C.; Zeuzem, S. J. Gastroenterol. Hepatol. Rev. 2010, 6, 70–77.
Return to citation in text: [1] -
Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Baker, A.; Earnshaw, D. L.; Hofmann, G. A.; Keenan, R. M.; Dhanak, D. Bioorg. Med. Chem. Lett. 2005, 15, 1553–1556. doi:10.1016/j.bmcl.2005.01.076
Return to citation in text: [1] [2] -
Swan, T. Hepatitis C: New Treatments in the Pipeline; Treatment Action Group: New York, April, 2008. http://www.treatmentactiongroup.org/assets/0/16/42/196/198/a8611995-345d-487f-89fd-f1d281c75040.pdf
Return to citation in text: [1] -
Pauwels, F.; Mostmans, W.; Quirynen, L. M. M.; van der Helm, L.; Boutton, C. W.; Rueff, A.-S.; Cleiren, E.; Raboisson, P.; Surleraux, D.; Nyanguile, O.; Simmen, K. A. J. Virol. 2007, 81, 6909–6919. doi:10.1128/JVI.01543-06
The potent activity of these series of products was correlated with the binding site identification and genotypic profiling of HCV polymerase inhibitors.
Return to citation in text: [1] -
Nájera, C.; Sansano, J. M. Org. Biomol. Chem. 2009, 7, 4567–4581. doi:10.1039/b913066g
Return to citation in text: [1] -
Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Hofmann, G. A.; Slater, M. J.; Haigh, D.; Dhanak, D.; Johnson, V. K.; Parry, N. R.; Thomes, P. Bioorg. Med. Chem. Lett. 2007, 17, 1930–1933. doi:10.1016/j.bmcl.2007.01.034
Return to citation in text: [1] [2] [3] -
Slater, M. J.; Amphlett, E. M.; Andrews, D. M.; Bravi, G.; Burton, G.; Cheasty, A. G.; Corfield, J. A.; Ellis, M. R.; Fenwick, R. H.; Fernandes, S.; Guidetti, R.; Haigh, D.; Hartley, C. D.; Howes, P. D.; Jackson, D. L.; Jarvest, R. L.; Lovegrove, V. L. H.; Medhurst, K. J.; Parry, N. R.; Price, H.; Shah, P.; Singh, O. M. P.; Stocker, R.; Thommes, P.; Wilkinson, C.; Wonacott, A. J. Med. Chem. 2007, 50, 897–900. doi:10.1021/jm061207r
Return to citation in text: [1] [2] -
Agbodjan, A. A.; Cooley, B. E.; Copley, R. C. B.; Corfield, J. A.; Flanagan, R. C.; Glover, B. N.; Guidetti, R.; Haigh, D.; Howes, P. D.; Jackson, M. M.; Matsuoka, R. T.; Medhurst, K. J.; Millar, A.; Sharp, M. J.; Slater, M. J.; Toczko, J. F.; Xie, S. J. Org. Chem. 2008, 73, 3094–3102. doi:10.1021/jo800062c
Return to citation in text: [1] [2] -
Flanagan, R. C.; Xie, S.; Millar, A. Org. Process Res. Dev. 2008, 12, 1307–1312. doi:10.1021/op8001799
Return to citation in text: [1] -
Haigh, D.; Amphlett, E. M.; Bravi, G. S.; Bright, H.; Chung, V.; Chambers, C. L.; Cheasty, A. G.; Convey, M. A.; Maire, A.; Ellis, M. R.; Fenwick, R.; Gray, D. F.; Hartley, C. D.; Howes, P. D.; Jarvest, R. L.; Medhurst, K. J.; Mehbob, A.; Mesogiti, D.; Mirzai, F.; Nerozzi, F.; Parry, N. R.; Roughley, N. R.; Skarynzski, T.; Slater, M. J.; Smith, S. A.; Stocker, R.; Theobald, C. J.; Thomas, P. J.; Thommes, P. A.; Thorpe, J. H.; Wilkinson, C. S.; Williams, E. W.
Identification of GSK625433: A novel clinical candidate for the treatment of hepatitis C.
233rd ACS National Meeting, Chicago, IL, March 25-29, 2007, Division of Medicinal Chemistry, First Time Disclosure of Clinical Candidates.
Return to citation in text: [1] -
Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Towards Heterocycles and Natural Products. In Chemistry of Heterocyclic Compounds; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons, Inc.: New Jersey, 2003; Vol. 59.
The potent activity of these series of products was correlated with the binding site identification and genotypic profiling of HCV polymerase inhibitors.
Return to citation in text: [1] -
Nájera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594
Return to citation in text: [1] -
Nair, V.; Suja, T. D. Tetrahedron 2007, 63, 12247–12275. doi:10.1016/j.tet.2007.09.065
see for recent review.
Return to citation in text: [1] -
Padwa, A.; Bur, S. K. Tetrahedron 2007, 63, 5341–5378. doi:10.1016/j.tet.2007.03.158
see for recent review.
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2007, 63, 3235–3285. doi:10.1016/j.tet.2007.01.009
see for recent review.
Return to citation in text: [1] -
Nájera, C.; Retamosa, M. G.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2007, 5038–5049. doi:10.1002/ejoc.200700267
Return to citation in text: [1] -
Nájera, C.; Sansano, J. M. Enantioselective Cycloadditions of Azomethine Ylides. In Synthesis of Heterocycles via Cycloaddition I; Hassner, A., Ed.; Topics in Heterocyclic Chemistry, Vol. 12; Springer, 2008; pp 117–145.
Return to citation in text: [1] -
Stanley, L. M.; Sibi, M. P. Chem. Rev. 2008, 108, 2887–2902. doi:10.1021/cr078371m
Return to citation in text: [1] -
Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Rev. 2008, 108, 3174–3198. doi:10.1021/cr078361l
Return to citation in text: [1] -
Naodovic, M.; Yamamoto, H. Chem. Rev. 2008, 108, 3132–3148. doi:10.1021/cr068413r
Return to citation in text: [1] -
Nájera, C.; Sansano, J. M.; Yus, M. J. Braz. Chem. Soc. 2010, 21, 377–412. doi:10.1590/S0103-50532010000300002
Return to citation in text: [1] [2] -
Nájera, C.; Retamosa, M. G.; Martín-Rodríguez, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2009, 5622–5634. doi:10.1002/ejoc.200900774
Return to citation in text: [1] [2] [3] [4] [5] -
Nájera, C.; Retamosa, M. G.; Sansano, J. M. Spanish Pat. Appl. P200800908, April 2, 2008.
Return to citation in text: [1] [2] [3] -
Tsubogo, T.; Saito, S.; Seki, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 13321–13332. doi:10.1021/ja8032058
Return to citation in text: [1] [2] -
Saito, S.; Tsubogo, T.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 5364–5365. doi:10.1021/ja0709730
Return to citation in text: [1] [2] [3] -
Nájera, C.; Retamosa, M. G.; Sansano, J. M. Angew. Chem., Int. Ed. 2008, 47, 6055–6058. doi:10.1002/anie.200801690
Return to citation in text: [1] [2] [3] -
Nájera, C.; Retamosa, M. G.; Sansano, J. M. Org. Lett. 2007, 9, 4025–4028. doi:10.1021/ol701577k
Return to citation in text: [1] [2] -
Nájera, C.; Retamosa, M. G.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Tetrahedron: Asymmetry 2008, 19, 2913–2923. doi:10.1016/j.tetasy.2008.12.021
Return to citation in text: [1] -
Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t
Return to citation in text: [1] -
Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045
Return to citation in text: [1] [2] -
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Wu, F. L. Tetrahedron: Asymmetry 2010, 21, 1184–1186. doi:10.1016/j.tetasy.2010.10.005
Return to citation in text: [1] [2] -
Teichert, J. F.; Feringa, B. L. Angew. Chem., Int. Ed. 2010, 49, 2486–2528. doi:10.1002/anie.200904948
Return to citation in text: [1] -
Alonso, I.; Trillo, B.; López, F.; Montserrat, S.; Ujaque, G.; Castedo, L.; Lledós, A.; Mascareñas, J. L. J. Am. Chem. Soc. 2009, 131, 13020–13030. doi:10.1021/ja905415r
Return to citation in text: [1] [2] [3] -
Teller, H.; Flügge, S.; Goddard, R.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 1949–1953. doi:10.1002/anie.200906550
Return to citation in text: [1] [2] -
González, A. Z.; Benítez, D.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 5500–5507. doi:10.1021/ja200084a
Return to citation in text: [1] [2] -
Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. Z. Naturforsch. 2009, 64b, 1469–1477.
Return to citation in text: [1] [2] -
de Cózar, A.; Cossío, F. P. Phys. Chem. Chem. Phys. 2011, 13, 10858–10868. doi:10.1039/C1CP20682F
Return to citation in text: [1] -
Svensson, M.; Humbel, S.; Morokuma, K. J. Chem. Phys. 1996, 105, 3654–3661. doi:10.1063/1.472235
Return to citation in text: [1] -
Vreven, T.; Morokuma, K. J. Comput. Chem. 2000, 21, 1419–1432. doi:10.1002/1096-987X(200012)21:16<1419::AID-JCC1>3.0.CO;2-C
Return to citation in text: [1] -
Dapprich, S.; Komáromi, I.; Byun, K. S.; Morokuma, K.; Frisch, M. J. J. Mol. Struct. (Theochem) 1999, 461-462, 1–21. doi:10.1016/S0166-1280(98)00475-8
Return to citation in text: [1] -
Gaussian09, Revision A.02; Gaussian Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Return to citation in text: [1] -
Kohn, W.; Becke, A. D.; Parr, R. G. J. Phys. Chem. 1996, 100, 12974–12980. doi:10.1021/jp960669l
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] -
Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200–1211. doi:10.1139/p80-159
Return to citation in text: [1] -
Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001
Return to citation in text: [1] -
Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299–303. doi:10.1063/1.448975
Return to citation in text: [1] -
Rappé, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A., III; Skiff, W. M. J. Am. Chem. Soc. 1992, 114, 10024–10035. doi:10.1021/ja00051a040
Return to citation in text: [1]
| 36. | Alonso, I.; Trillo, B.; López, F.; Montserrat, S.; Ujaque, G.; Castedo, L.; Lledós, A.; Mascareñas, J. L. J. Am. Chem. Soc. 2009, 131, 13020–13030. doi:10.1021/ja905415r |
| 37. | Teller, H.; Flügge, S.; Goddard, R.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 1949–1953. doi:10.1002/anie.200906550 |
| 38. | González, A. Z.; Benítez, D.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 5500–5507. doi:10.1021/ja200084a |
| 39. | Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. Z. Naturforsch. 2009, 64b, 1469–1477. |
| 1. | Gao, M.; Nettles, R. E.; Belema, M.; Snyder, L. B.; Nguyen, V. N.; Fridell, R. A.; Serrano-Wu, M. H.; Langley, D. R.; Sun, J.-H.; O’Boyle, D. R., II; Lemm, J. A.; Wang, C.; Knipe, J. O.; Chien, C.; Colonno, R. J.; Grasela, D. M.; Meanwell, N. A.; Hamann, L. G. Nature 2010, 465, 96–100. doi:10.1038/nature08960 |
| 6. | Swan, T. Hepatitis C: New Treatments in the Pipeline; Treatment Action Group: New York, April, 2008. http://www.treatmentactiongroup.org/assets/0/16/42/196/198/a8611995-345d-487f-89fd-f1d281c75040.pdf |
| 7. |
Pauwels, F.; Mostmans, W.; Quirynen, L. M. M.; van der Helm, L.; Boutton, C. W.; Rueff, A.-S.; Cleiren, E.; Raboisson, P.; Surleraux, D.; Nyanguile, O.; Simmen, K. A. J. Virol. 2007, 81, 6909–6919. doi:10.1128/JVI.01543-06
The potent activity of these series of products was correlated with the binding site identification and genotypic profiling of HCV polymerase inhibitors. |
| 11. | Agbodjan, A. A.; Cooley, B. E.; Copley, R. C. B.; Corfield, J. A.; Flanagan, R. C.; Glover, B. N.; Guidetti, R.; Haigh, D.; Howes, P. D.; Jackson, M. M.; Matsuoka, R. T.; Medhurst, K. J.; Millar, A.; Sharp, M. J.; Slater, M. J.; Toczko, J. F.; Xie, S. J. Org. Chem. 2008, 73, 3094–3102. doi:10.1021/jo800062c |
| 45. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 46. | Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098 |
| 47. | Kohn, W.; Becke, A. D.; Parr, R. G. J. Phys. Chem. 1996, 100, 12974–12980. doi:10.1021/jp960669l |
| 48. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 49. | Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200–1211. doi:10.1139/p80-159 |
| 50. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 5. | Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Baker, A.; Earnshaw, D. L.; Hofmann, G. A.; Keenan, R. M.; Dhanak, D. Bioorg. Med. Chem. Lett. 2005, 15, 1553–1556. doi:10.1016/j.bmcl.2005.01.076 |
| 27. | Tsubogo, T.; Saito, S.; Seki, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 13321–13332. doi:10.1021/ja8032058 |
| 28. | Saito, S.; Tsubogo, T.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 5364–5365. doi:10.1021/ja0709730 |
| 51. | Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299–303. doi:10.1063/1.448975 |
| 1. | Gao, M.; Nettles, R. E.; Belema, M.; Snyder, L. B.; Nguyen, V. N.; Fridell, R. A.; Serrano-Wu, M. H.; Langley, D. R.; Sun, J.-H.; O’Boyle, D. R., II; Lemm, J. A.; Wang, C.; Knipe, J. O.; Chien, C.; Colonno, R. J.; Grasela, D. M.; Meanwell, N. A.; Hamann, L. G. Nature 2010, 465, 96–100. doi:10.1038/nature08960 |
| 4. | Lange, C. M.; Sarrazin, C.; Zeuzem, S. J. Gastroenterol. Hepatol. Rev. 2010, 6, 70–77. |
| 25. | Nájera, C.; Retamosa, M. G.; Martín-Rodríguez, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2009, 5622–5634. doi:10.1002/ejoc.200900774 |
| 26. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Spanish Pat. Appl. P200800908, April 2, 2008. |
| 41. | Svensson, M.; Humbel, S.; Morokuma, K. J. Chem. Phys. 1996, 105, 3654–3661. doi:10.1063/1.472235 |
| 42. | Vreven, T.; Morokuma, K. J. Comput. Chem. 2000, 21, 1419–1432. doi:10.1002/1096-987X(200012)21:16<1419::AID-JCC1>3.0.CO;2-C |
| 43. | Dapprich, S.; Komáromi, I.; Byun, K. S.; Morokuma, K.; Frisch, M. J. J. Mol. Struct. (Theochem) 1999, 461-462, 1–21. doi:10.1016/S0166-1280(98)00475-8 |
| 2. | Tellinghuisen, T. L.; Evans, M. J.; von Hahn, T.; You, S.; Rice, C. M. J. Virol. 2007, 81, 8853–8867. doi:10.1128/JVI.00753-07 |
| 3. | Gish, R. Future Therapies for Hepatitis C. In The HCV Advocate Medical Writers’ Circle; Franciscus, A., Ed.; HCSP Publications: San Francisco, February 1, 2006. |
| 27. | Tsubogo, T.; Saito, S.; Seki, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 13321–13332. doi:10.1021/ja8032058 |
| 28. | Saito, S.; Tsubogo, T.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 5364–5365. doi:10.1021/ja0709730 |
| 14. |
Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Towards Heterocycles and Natural Products. In Chemistry of Heterocyclic Compounds; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons, Inc.: New Jersey, 2003; Vol. 59.
The potent activity of these series of products was correlated with the binding site identification and genotypic profiling of HCV polymerase inhibitors. |
| 15. | Nájera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594 |
| 16. |
Nair, V.; Suja, T. D. Tetrahedron 2007, 63, 12247–12275. doi:10.1016/j.tet.2007.09.065
see for recent review. |
| 17. |
Padwa, A.; Bur, S. K. Tetrahedron 2007, 63, 5341–5378. doi:10.1016/j.tet.2007.03.158
see for recent review. |
| 18. |
Pellissier, H. Tetrahedron 2007, 63, 3235–3285. doi:10.1016/j.tet.2007.01.009
see for recent review. |
| 19. | Nájera, C.; Retamosa, M. G.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2007, 5038–5049. doi:10.1002/ejoc.200700267 |
| 40. | de Cózar, A.; Cossío, F. P. Phys. Chem. Chem. Phys. 2011, 13, 10858–10868. doi:10.1039/C1CP20682F |
| 13. |
Haigh, D.; Amphlett, E. M.; Bravi, G. S.; Bright, H.; Chung, V.; Chambers, C. L.; Cheasty, A. G.; Convey, M. A.; Maire, A.; Ellis, M. R.; Fenwick, R.; Gray, D. F.; Hartley, C. D.; Howes, P. D.; Jarvest, R. L.; Medhurst, K. J.; Mehbob, A.; Mesogiti, D.; Mirzai, F.; Nerozzi, F.; Parry, N. R.; Roughley, N. R.; Skarynzski, T.; Slater, M. J.; Smith, S. A.; Stocker, R.; Theobald, C. J.; Thomas, P. J.; Thommes, P. A.; Thorpe, J. H.; Wilkinson, C. S.; Williams, E. W.
Identification of GSK625433: A novel clinical candidate for the treatment of hepatitis C. 233rd ACS National Meeting, Chicago, IL, March 25-29, 2007, Division of Medicinal Chemistry, First Time Disclosure of Clinical Candidates. |
| 20. | Nájera, C.; Sansano, J. M. Enantioselective Cycloadditions of Azomethine Ylides. In Synthesis of Heterocycles via Cycloaddition I; Hassner, A., Ed.; Topics in Heterocyclic Chemistry, Vol. 12; Springer, 2008; pp 117–145. |
| 21. | Stanley, L. M.; Sibi, M. P. Chem. Rev. 2008, 108, 2887–2902. doi:10.1021/cr078371m |
| 22. | Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Rev. 2008, 108, 3174–3198. doi:10.1021/cr078361l |
| 23. | Naodovic, M.; Yamamoto, H. Chem. Rev. 2008, 108, 3132–3148. doi:10.1021/cr068413r |
| 24. | Nájera, C.; Sansano, J. M.; Yus, M. J. Braz. Chem. Soc. 2010, 21, 377–412. doi:10.1590/S0103-50532010000300002 |
| 39. | Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. Z. Naturforsch. 2009, 64b, 1469–1477. |
| 9. | Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Hofmann, G. A.; Slater, M. J.; Haigh, D.; Dhanak, D.; Johnson, V. K.; Parry, N. R.; Thomes, P. Bioorg. Med. Chem. Lett. 2007, 17, 1930–1933. doi:10.1016/j.bmcl.2007.01.034 |
| 10. | Slater, M. J.; Amphlett, E. M.; Andrews, D. M.; Bravi, G.; Burton, G.; Cheasty, A. G.; Corfield, J. A.; Ellis, M. R.; Fenwick, R. H.; Fernandes, S.; Guidetti, R.; Haigh, D.; Hartley, C. D.; Howes, P. D.; Jackson, D. L.; Jarvest, R. L.; Lovegrove, V. L. H.; Medhurst, K. J.; Parry, N. R.; Price, H.; Shah, P.; Singh, O. M. P.; Stocker, R.; Thommes, P.; Wilkinson, C.; Wonacott, A. J. Med. Chem. 2007, 50, 897–900. doi:10.1021/jm061207r |
| 11. | Agbodjan, A. A.; Cooley, B. E.; Copley, R. C. B.; Corfield, J. A.; Flanagan, R. C.; Glover, B. N.; Guidetti, R.; Haigh, D.; Howes, P. D.; Jackson, M. M.; Matsuoka, R. T.; Medhurst, K. J.; Millar, A.; Sharp, M. J.; Slater, M. J.; Toczko, J. F.; Xie, S. J. Org. Chem. 2008, 73, 3094–3102. doi:10.1021/jo800062c |
| 12. | Flanagan, R. C.; Xie, S.; Millar, A. Org. Process Res. Dev. 2008, 12, 1307–1312. doi:10.1021/op8001799 |
| 34. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Wu, F. L. Tetrahedron: Asymmetry 2010, 21, 1184–1186. doi:10.1016/j.tetasy.2010.10.005 |
| 5. | Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Baker, A.; Earnshaw, D. L.; Hofmann, G. A.; Keenan, R. M.; Dhanak, D. Bioorg. Med. Chem. Lett. 2005, 15, 1553–1556. doi:10.1016/j.bmcl.2005.01.076 |
| 8. | Nájera, C.; Sansano, J. M. Org. Biomol. Chem. 2009, 7, 4567–4581. doi:10.1039/b913066g |
| 9. | Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Hofmann, G. A.; Slater, M. J.; Haigh, D.; Dhanak, D.; Johnson, V. K.; Parry, N. R.; Thomes, P. Bioorg. Med. Chem. Lett. 2007, 17, 1930–1933. doi:10.1016/j.bmcl.2007.01.034 |
| 9. | Burton, G.; Ku, T. W.; Carr, T. J.; Kiesow, T.; Sarisky, R. T.; Lin-Goerke, J.; Hofmann, G. A.; Slater, M. J.; Haigh, D.; Dhanak, D.; Johnson, V. K.; Parry, N. R.; Thomes, P. Bioorg. Med. Chem. Lett. 2007, 17, 1930–1933. doi:10.1016/j.bmcl.2007.01.034 |
| 10. | Slater, M. J.; Amphlett, E. M.; Andrews, D. M.; Bravi, G.; Burton, G.; Cheasty, A. G.; Corfield, J. A.; Ellis, M. R.; Fenwick, R. H.; Fernandes, S.; Guidetti, R.; Haigh, D.; Hartley, C. D.; Howes, P. D.; Jackson, D. L.; Jarvest, R. L.; Lovegrove, V. L. H.; Medhurst, K. J.; Parry, N. R.; Price, H.; Shah, P.; Singh, O. M. P.; Stocker, R.; Thommes, P.; Wilkinson, C.; Wonacott, A. J. Med. Chem. 2007, 50, 897–900. doi:10.1021/jm061207r |
| 25. | Nájera, C.; Retamosa, M. G.; Martín-Rodríguez, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2009, 5622–5634. doi:10.1002/ejoc.200900774 |
| 24. | Nájera, C.; Sansano, J. M.; Yus, M. J. Braz. Chem. Soc. 2010, 21, 377–412. doi:10.1590/S0103-50532010000300002 |
| 25. | Nájera, C.; Retamosa, M. G.; Martín-Rodríguez, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2009, 5622–5634. doi:10.1002/ejoc.200900774 |
| 26. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Spanish Pat. Appl. P200800908, April 2, 2008. |
| 29. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Angew. Chem., Int. Ed. 2008, 47, 6055–6058. doi:10.1002/anie.200801690 |
| 52. | Rappé, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A., III; Skiff, W. M. J. Am. Chem. Soc. 1992, 114, 10024–10035. doi:10.1021/ja00051a040 |
| 30. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Org. Lett. 2007, 9, 4025–4028. doi:10.1021/ol701577k |
| 31. | Nájera, C.; Retamosa, M. G.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Tetrahedron: Asymmetry 2008, 19, 2913–2923. doi:10.1016/j.tetasy.2008.12.021 |
| 36. | Alonso, I.; Trillo, B.; López, F.; Montserrat, S.; Ujaque, G.; Castedo, L.; Lledós, A.; Mascareñas, J. L. J. Am. Chem. Soc. 2009, 131, 13020–13030. doi:10.1021/ja905415r |
| 37. | Teller, H.; Flügge, S.; Goddard, R.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 1949–1953. doi:10.1002/anie.200906550 |
| 38. | González, A. Z.; Benítez, D.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 5500–5507. doi:10.1021/ja200084a |
| 25. | Nájera, C.; Retamosa, M. G.; Martín-Rodríguez, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2009, 5622–5634. doi:10.1002/ejoc.200900774 |
| 29. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Angew. Chem., Int. Ed. 2008, 47, 6055–6058. doi:10.1002/anie.200801690 |
| 25. | Nájera, C.; Retamosa, M. G.; Martín-Rodríguez, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2009, 5622–5634. doi:10.1002/ejoc.200900774 |
| 26. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Spanish Pat. Appl. P200800908, April 2, 2008. |
| 28. | Saito, S.; Tsubogo, T.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 5364–5365. doi:10.1021/ja0709730 |
| 29. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Angew. Chem., Int. Ed. 2008, 47, 6055–6058. doi:10.1002/anie.200801690 |
| 30. | Nájera, C.; Retamosa, M. G.; Sansano, J. M. Org. Lett. 2007, 9, 4025–4028. doi:10.1021/ol701577k |
| 33. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 35. | Teichert, J. F.; Feringa, B. L. Angew. Chem., Int. Ed. 2010, 49, 2486–2528. doi:10.1002/anie.200904948 |
| 36. | Alonso, I.; Trillo, B.; López, F.; Montserrat, S.; Ujaque, G.; Castedo, L.; Lledós, A.; Mascareñas, J. L. J. Am. Chem. Soc. 2009, 131, 13020–13030. doi:10.1021/ja905415r |
| 32. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 33. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 34. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Wu, F. L. Tetrahedron: Asymmetry 2010, 21, 1184–1186. doi:10.1016/j.tetasy.2010.10.005 |
© 2011 Martín-Rodríguez et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)