Abstract
A convenient synthesis of the tetrasaccharide repeating unit of the O-antigen of Shigella boydii type 9 has been achieved in excellent yield using a [2 + 2] block glycosylation strategy. TEMPO-mediated selective oxidation of the primary alcohol of the tetrasaccharide derivative 8 to the carboxylic group followed by deprotection of the functional groups furnished target tetrasaccharide 1 as its 4-methoxyphenyl glycoside in high yield.

Graphical Abstract
Introduction
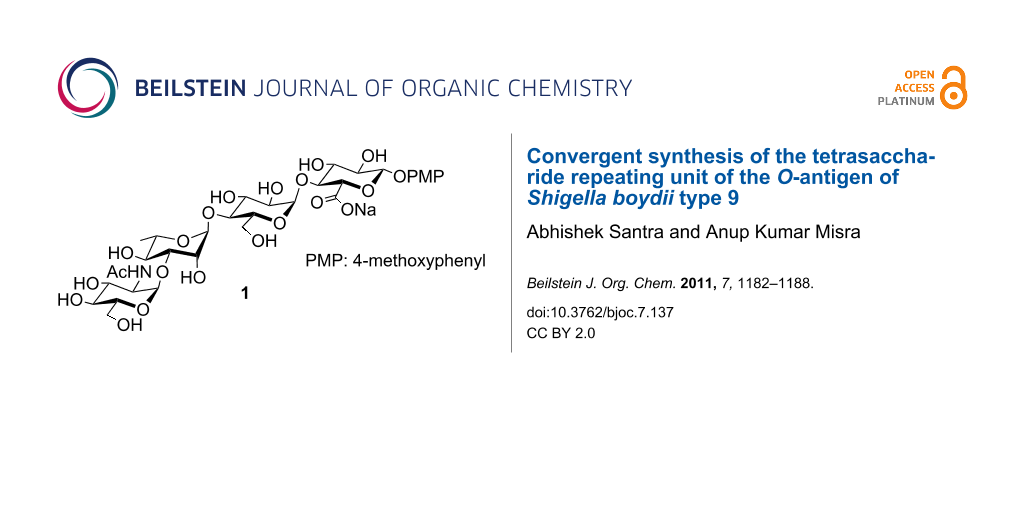
Diarrhoeal disease is a common cause of death in the tropical countries and it is the second mostly causing infant deaths worldwide. Shigella is one of the well-studied human pathogens that cause diarrhoeal disease and dysentery (e.g., shigellosis). Among several types of Shigella species, Shigella dysenteriae is the most virulent pathogen causing devastating health problems in developing countries [1-3]. Shigella strains are classified into four species: Shigella boydii, Shigella dysenteriae, Shigella flexneri and Shigella sonnei [4]. Sometimes, these species are also termed as Shigella subgroups A, B, C, and D. Based on the O-antigens, the Shigella species are divided into multiple serotypes [5]. In general, O-antigens of Shigella species are acidic in nature because of the presence of acidic constituents (e.g., uronic acid, pseudaminic acid etc. or lactic acid, pyruvic acid etc.) in their structures [6,7]. Recently, L’vov et al. reported the structure of the O-antigen of Shigella boydii type 9, which is a tetrasaccharide repeating unit containing a D-glucuronic acid moiety (Figure 1) [8].
![[1860-5397-7-137-1]](/bjoc/content/figures/1860-5397-7-137-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the tetrasaccharide repeating unit of the O-antigen of Shigella boydii type 9.
Figure 1: Structure of the tetrasaccharide repeating unit of the O-antigen of Shigella boydii type 9.
Development of effective therapeutics to control the infections of drug-resistant bacterial strains is the thrust area in the medicinal chemistry. Like other bacterial infections, emergence of the drug resistant Shigella infections requires development of the newer therapeutics than the earlier used anti-shigellosis agents [9,10]. Because of the high antigenic nature of the O-antigens, antibodies against the O-specific polysaccharide of a particular Shigella strain have the potential to control Shigella infections [11-14]. A number of reports have been cited earlier to develop glycoconjugate based therapy to control Shigella infections [15-19]. In order to develop a glycoconjugate based therapeutic agent from the tetrasaccharide repeating unit of the O-antigen of Shigella boydii type 9, it is essential to perform several immunochemical studies with the glycoconjugates derived from this tetrasaccharide repeating unit. The large quantity of the tetrasaccharide that is required for this purpose cannot be accessible from a natural source. Therefore, chemical synthesis is the only option for achieving this in large quantity. As a first step towards the preparation of glycoconjugates, we report herein a convergent chemical synthesis of the tetrasaccharide as its 4-methoxyphenyl glycoside 1 corresponding to the O-antigen of Shigella boydii type 9 using a [2 + 2] block glycosylation strategy (Figure 2).
![[1860-5397-7-137-2]](/bjoc/content/figures/1860-5397-7-137-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of the synthesized tetrasaccharide and its intermediates.
Figure 2: Structure of the synthesized tetrasaccharide and its intermediates.
Results and Discussion
The convergent synthesis of the target tetrasaccharide 1 as its 4-methoxyphenyl glycoside has been achieved applying a number of recently developed elegant reaction methodologies. A number of notable features are present in the synthetic strategy, which are (a) convergent [2 + 2] block glycosylation; (b) application of recently developed environmentally benign reaction conditions for protecting group manipulations and glycosylations such as, (i) O-acetylation using sulfamic acid [20], (ii) regioselective ring opening of the benzylidene acetal using a combination of triethylsilane and iodine [21], (iii) direct one-pot conversion of O-acetyl group to O-benzyl group [22], (iv) activation of glycosyl trichloroacetimidate and thioglycoside donors by perchloric acid supported on silica (HClO4–SiO2) [23-26], and late stage TEMPO mediated selective oxidation [27-29] of the primary hydroxy group to the carboxylic group under phase transfer conditions without affecting the secondary hydroxy groups; (c) use of 4-methoxyphenyl (PMP) group as anomeric protecting group [30,31], which can be easily removed under oxidative conditions for the preparation of glycoconjugate derivatives. The synthesis of the target tetrasaccharide 1 was achieved by the stereoselective coupling of a D-maltose derived disaccharide derivative 2 and a disaccharide thioglycoside derivative 7 followed by functional group manipulations of the resulting tetrasaccharide derivative 8. For this purpose, suitably functionalized reaction intermediates 2, 3, 4 and 7 were prepared from commercially available reducing mono- and disaccharides utilizing a series of reaction methodologies reported earlier.
4-Methoxyphenyl (4,6-O-benzylidene-α-D-glucopyranosyl)-(1→4)-β-D-glucopyranoside (5) [32] was subjected to a sequence of reactions involving acetylation using acetic anhydride in the presence of sulfamic acid [20] followed by regioselective reductive ring opening of the benzylidene acetal using a combination of triethylsilane and iodine [21] to furnish 4-methoxyphenyl (2,3-di-O-acetyl-6-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-acetyl-β-D-glucopyranoside (2) in 82% yield (Scheme 1). 3,4,6-Tri-O-acetyl-2-azido-2-deoxy-α-D-glucopyranosyl trichloroacetimidate (3) [33] was allowed to couple stereoselectively with ethyl 2,4-di-O-benzyl-1-thio-α-L-rhamnopyranoside (4) [34] under Schmidt’s reaction conditions [35] using perchloric acid supported on silica (HClO4–SiO2) [23,24] as glycosylation activator to give ethyl (3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-D-glucopyranosyl)-(1→3)-2,4-di-O-benzyl-1-thio-α-L-rhamnopyranoside (6) in 81% yield. Stereoselective formation of compound 6 was confirmed from its spectral analysis (presence of signals at δ 5.35 (br s, H-1C), 4.95 (d, J = 3.6 Hz, H-1D) in the 1H NMR and signals at δ 92.8 (C-1D), 81.0 (C-1C) in the 13C NMR spectrum). Compound 6 was transformed into disaccharide thioglycoside donor 7 in 91% yield under a one-pot deacetylation–benzylation reaction condition [22] (Scheme 2). In this case, the thioethyl group acts as an orthogonal anomeric protecting group since it acts as a glycosyl acceptor in the case of compound 4 whereas compound 7 has been used as the glycosyl donor in the next step. Iodonium ion promoted stereoselective glycosylation of the disaccharide thioglycoside donor 7 with the disaccharide acceptor 2 in the presence of a combination of N-iodosuccinimide (NIS) and HClO4–SiO2 [25,26] furnished tetrasaccharide derivative 8 in 82% yield. Stereoselective formation of new α-glycosyl linkage in compound 8 was confirmed from the 1D and 2D NMR spectral analysis (presence of signals at δ 5.34 (d, J = 4.5 Hz, H-1B), 4.97 (d, J = 3.5 Hz, H-1D), 4.95 (d, J = 8.0 Hz, H-1A), 4.89 (d, J = 2.0 Hz, H-1C) in the 1H NMR and signals at δ 99.7 (C-1A), 98.8 (C-1C), 96.0 (C-1B), 93.9 (C-1D) in the 13C NMR spectrum). Because of the presence of the non-participating benzyloxy group in the C-2 position of the L-rhamnosyl moiety, involved in the glycosylation reaction, a minor amount of β-glycosyl linked product also formed (~6%) together with the desired product 8, which was separated by the column chromatography. The presence of the α-linkages in compound 8 was further confirmed from the JC1-H1 values in the 1H coupled 13C NMR spectrum of compound 8. Appearance of JC1-H1 172.0 Hz, 171.6 Hz, 170.5 Hz and 162.0 Hz values in the anomeric region in the 1H coupled 13C spectrum unambiguously supported the presence of three α-linkages and one β-linkage [36-38] in compound 8. Compound 8 was subjected to a reaction sequence involving (a) deacetylation using 0.1 M sodium methoxide in methanol; (b) TEMPO mediated selective oxidation [27-29] of the primary hydroxy group leaving secondary hydroxy groups unaffected in a phase transfer reaction condition and (c) removal of benzyl groups for cleavage of ethers and reduction of the azido group to an amine by hydrogenation over 20% Pd(OH)2/C followed by N-acetylation to furnish target tetrasaccharide 1 as its sodium salt and 4-methoxyphenyl glycoside in 64% yield (Scheme 3). Spectroscopic analysis of compound 1 confirmed its formation (presence of signals at δ 5.20 (d, J = 3.6 Hz, H-1B), 4.93 (br s, H-1D), 4.89 (br s, H-1C), 4.82 (d, J = 7.8 Hz, H-1A) in the 1H NMR and signals at δ 103.3 (C-1A), 102.8 (C-1B), 102.4 (C-1C), 96.7 (C-1D) in the 13C NMR spectrum).
![[1860-5397-7-137-i1]](/bjoc/content/inline/1860-5397-7-137-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents: (a) acetic anhydride, sulfamic acid, 60 °C, 30 min, 91%; (b) Et3SiH, I2, 0–5 °C, 30 min, 82%.
Scheme 1: Reagents: (a) acetic anhydride, sulfamic acid, 60 °C, 30 min, 91%; (b) Et3SiH, I2, 0–5 °C, 30 min, ...
![[1860-5397-7-137-i2]](/bjoc/content/inline/1860-5397-7-137-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents: (a) HClO4–SiO2, CH2Cl2, −15 °C, 1 h, 81%; (b) benzyl bromide, NaOH, n-Bu4NBr, THF, rt, 2 h, 91%.
Scheme 2: Reagents: (a) HClO4–SiO2, CH2Cl2, −15 °C, 1 h, 81%; (b) benzyl bromide, NaOH, n-Bu4NBr, THF, rt, 2 ...
![[1860-5397-7-137-i3]](/bjoc/content/inline/1860-5397-7-137-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents: (a) N-iodosuccinimide, HClO4–SiO2, −10 °C, 1 h, 82%; (b) 0.1 M CH3ONa, CH3OH, rt, 3 h; (c) (i) NaBr, TBAB, TEMPO, CH2Cl2, H2O, NaOCl, NaHCO3, 5 °C, 2 h; (ii) tert-butanol, 2-methylbut-2-ene, NaClO2, NaH2PO4, rt, 3 h; (d) (i) H2, 20% Pd(OH)2/C, CH3OH–EtOAc, rt, 10 h; (ii) acetic anhydride, CH3OH, rt, 30 min, overall 64%.
Scheme 3: Reagents: (a) N-iodosuccinimide, HClO4–SiO2, −10 °C, 1 h, 82%; (b) 0.1 M CH3ONa, CH3OH, rt, 3 h; (c...
Conclusion
In conclusion, a convenient synthetic strategy has been developed for the synthesis of the tetrasaccharide repeating unit of the O-antigen of Shigella boydii type 9 as its 4-methoxyphenyl glycoside sodium salt using a [2 + 2] block synthetic strategy. Use of a block glycosylation strategy and a late-stage selective oxidation of the primary hydroxy group significantly reduced the number of protection–deprotection steps. A number of modified clean reaction methodologies have been applied for the preparation of intermediates. HClO4–SiO2 has been used as an effective acid catalyst to activate glycosyl trichloroacetimidate derivative and thioglycoside in combination with NIS avoiding the use of moisture sensitive protic acids. All intermediate steps were high yielding and glycosylation steps were stereoselective.
Experimental
General methods: All reactions were monitored by thin layer chromatography over silica gel-coated TLC plates. The spots on TLC plates were visualized by warming ceric sulfate (2% Ce(SO4)2 in 2 N H2SO4)-sprayed plates on a hot plate. Silica gel 230–400 mesh was used for column chromatography. 1H and 13C NMR, DEPT 135, 2D COSY, HMQC and gated 1H coupled 13C NMR spectra were recorded on Bruker Avance DRX 500 and 600 MHz spectrometers using CDCl3 and CD3OD as solvents and TMS as internal standard unless otherwise stated. Chemical shifts are expressed in δ ppm. ESIMS were recorded on a Micromass Quattro mass spectrometer. Elementary analysis was carried out on Carlo Erba-1108 analyzer. Optical rotations were measured at 25 °C on a Jasco P-2000 polarimeter. Commercially available grades of organic solvents of adequate purity are used in all reactions. Silica supported perchloric acid (HClO4–SiO2) was prepared following the earlier report [39].
4-Methoxyphenyl (2,3-di-O-acetyl-6-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-acetyl-β-D-glucopyranoside (2): To a suspension of 4-methoxyphenyl (4,6-O-benzylidene-α-D-glucopyranosyl)-(1→4)-β-D-glucopyranoside (5, 3.0 g, 5.59 mmol) in acetic anhydride (6.0 mL, 63.47 mmol) sulfamic acid (100 mg, 1.0 mmol) was added and the reaction mixture was allowed to stir at 60 °C for 30 min. The solvents were removed under reduced pressure and the crude reaction mixture was passed through a short pad of silica gel using hexane–EtOAc (1:2) as eluant to give pure acetylated product (3.8 g, 91%). To a solution of the acetylated product (3.5 g, 4.69 mmol) in CH3CN (15 mL) triethylsilane (1.5 mL, 9.39 mmol) and iodine (250 mg, 0.98 mmol) were added at 0–5 °C and the reaction mixture was allowed to stir at the same temperature for 30 min. The reaction mixture was poured into water and extracted with CH2Cl2 (100 mL). The organic layer was successively washed with satd. NaHCO3 solution and water, dried (Na2SO4) and concentrated. The crude product was purified over silica gel using hexane–EtOAc (2:1) as eluant to give pure compound 2 (2.9 g, 82%). White solid; mp 60–62 °C; [α]D25 +43.2 (c 1.2, CHCl3); IR (KBr): 3479, 1751, 1509, 1371, 1233, 1047, 830, 754 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.38 (d, J = 4.0 Hz, 1H, H-1B), 5.28 (t, J = 9.0 Hz, 1H, H-3A), 5.21 (t, J = 9.0 Hz, 1H, H-3B), 5.04 (t, J = 8.5 Hz, 1H, H-2A), 4.96 (d, J = 8.0 Hz, 1H, H-1A), 4.80 (dd, J = 9.0, 4.0 Hz, 1H, H-2B), 4.60–4.54 (m, 2H, PhCH2), 4.50 (d, J = 11.5 Hz, 1H, H-6aA), 4.22 (dd, J = 12.0 Hz, 5.0 Hz, 1H, H-6bA), 4.06 (t, J = 9.5 Hz, 1H, H-4A), 3.79–3.71 (m, 4H, H-4B, H-5A, H-5B, H-6aB), 3.75 (s, 3H, OCH3), 3.63–3.61 (m, 1H, H-6bB), 2.07, 2.03, 2.00, 1.99 (4 s, 15H, 5 COCH3); 13C NMR (125 MHz, CDCl3) δ 170.1, 170.6, 170.5, 170.2, 169.6 (5 COCH3), 155.6–114.5 (Ar-C), 99.6 (C-1A), 95.7 (C-1B), 75.4 (C-3A), 73.7 (PhCH2), 72.3 (C-4A), 72.1 (C-3B), 72.0 (C-2A), 71.9 (C-5A), 71.5 (C-5B), 70.0 (C-2B), 69.7 (C-4B), 68.8 (C-6B), 62.7 (C-6A), 55.6 (OCH3), 20.8, 20.7, 20.6, 20.5 (2 C) (5 COCH3); ESIMS m/z: 771.2 [M + Na]+; anal. calcd for C36H44O17 (748.26): C, 57.75; H, 5.92; found: C, 57.54; H, 6.15.
Ethyl (3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-D-glucopyranosyl)-(1→3)-2,4-di-O-benzyl-1-thio-α-L-rhamnopyranoside (6): A solution of compound 3 (2.4 g, 5.04 mmol) and compound 4 (1.5 g, 3.86 mmol) in anhydrous CH2Cl2 (10 mL) was cooled to −15 °C under argon. To the cooled reaction mixture HClO4–SiO2 (50.0 mg) was added and the reaction mixture was allowed to stir at the same temperature for 1 h. The reaction mixture was filtered through a bed of Celite® and concentrated under reduced pressure. The crude product was purified over silica gel using hexane–EtOAc (3:1) as eluant to furnish pure compound 6 (2.2 g, 81%). Yellow oil; [α]D25 +63.3 (c 1.2, CHCl3); IR (neat): 2930, 2110, 1750, 1455, 1368, 1233, 1094, 1039, 755, 699 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.43–7.26 (m, 10H, Ar-H), 5.55 (t, J = 10.2 Hz, 1H, H-3D), 5.35 (br s, 1H, H-1C), 4.98 (t, J = 10.2 Hz, 1H, H-4D), 4.95 (d, J = 3.6 Hz, 1H, H-1D), 4.91 (d, J = 11.4 Hz, 1H, PhCH2), 4.74 (d, J = 12.0 Hz, 1H, PhCH2), 4.70 (d, J = 11.4 Hz, 1H, PhCH2), 4.64 (d, J = 12.0 Hz, 1H, PhCH2), 4.15–4.13 (m, 1H, H-5D), 4.08–4.03 (m, 2H, H-5C, H-6aD), 4.00 (dd, J = 9.6, 3.0 Hz, 1H, H-3C), 3.93 (br s, 1H, H-2C), 3.88 (d, J = 12.0 Hz, 1H, H-6bD), 3.69 (t, J = 9.6 Hz, 1H, H-4C), 3.31 (dd, J = 10.8, 3.6 Hz, 1H, H-2D), 2.61–2.56 (m, 2H, SCH2CH3), 2.08, 2.05, 1.90 (3 s, 9H, 3 COCH3), 1.36 (d, J = 6.0 Hz, 3H, CCH3), 1.25 (t, J = 7.2 Hz, 3H, SCH2CH3); 13C NMR (150 MHz, CDCl3) δ 170.5, 169.8, 169.6 (3 COCH3), 138.0–127.6 (Ar-C), 92.8 (C-1D), 81.0 (C-1C), 79.4 (C-3C), 75.5 (PhCH2), 74.6 (C-3C), 74.2 (C-2C), 71.8 (PhCH2), 70.4 (C-3D), 68.4 (C-5C), 68.1 (C-4D), 67.3 (C-5D), 61.5 (C-6D), 60.6 (C-2D), 25.4 (SCH2CH3), 20.7, 20.6, 20.5 (3 COCH3), 17.7 (CCH3), 14.9 (SCH2CH3); ESIMS m/z: 724.2 [M + Na]+; anal. calcd for C34H43N3O11S (701.26): C, 58.19; H, 6.18; found: C, 58.0; H, 6.42.
Ethyl (2-azido-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl)-(1→3)-2,4-di-O-benzyl-1-thio-α-L-rhamnopyranoside (7): To a solution of compound 6 (2.0 g, 2.85 mmol) in THF (10 mL) powdered NaOH (1.0 g, 25 mmol), benzyl bromide (2.1 mL, 17.6 mmol) and n-Bu4NBr (50 mg) were added and the reaction mixture was allowed to stir at rt for 2 h. The reaction mixture was poured into water and extracted with CH2Cl2 (100 mL). The organic layer was washed with water, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over silica gel using hexane–EtOAc (6:1) as eluant to give pure compound 7 (2.2 g, 91%). Yellow oil; [α]D25 +12.0 (c 1.2, CHCl3); IR (neat): 3430, 3031, 2925, 2107, 1642, 1496, 1454, 1361, 1215, 1091, 1051, 1028, 751, 697 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.45–7.07 (m, 25H, Ar-H), 5.32 (br s, 1H, H-1C), 4.93 (d, J = 3.6 Hz, 1H, H-1D), 4.85 (br s, 2H, PhCH2), 4.83 (d, J = 10.2 Hz, 1H, PhCH2), 4.77 (d, J = 10.8 Hz, 1H, PhCH2), 4.74 (d, J = 12.0 Hz, 1H, PhCH2), 4.69 (d, J = 12.0 Hz, 1H, PhCH2), 4.60 (d, J = 12.6 Hz, 1H, PhCH2), 4.55 (d, J = 10.2 Hz, 1H, PhCH2), 4.48 (d, J = 10.8 Hz, 1H, PhCH2), 4.35 (d, J = 12.6 Hz, 1H, PhCH2), 4.07 (t, J = 9.6 Hz, 1H, H-3D), 4.04–3.99 (m, 3H, H-3C, H-5C, H-5D), 3.93 (br s, 1H, H-2C), 3.79 (t, J = 9.6 Hz, 1H, H-4C), 3.66 (t, J = 9.6 Hz, 1H, H-4D), 3.60 (dd, J = 10.8, 2.4 Hz, 1H, H-6aD), 3.51 (dd, J = 10.8, 1.2 Hz, 1H, H-6aD), 3.40 (dd, J = 10.2, 3.6 Hz, 1H, H-2D), 2.60–2.50 (m, 2H, SCH2CH3), 1.35 (d, J = 6.0 Hz, 3H, CCH3), 1.22 (t, J = 7.8 Hz, 3H, SCH2CH3); 13C NMR (150 MHz, CDCl3) δ 137.7–127.5 (Ar-C), 93.2 (C-1D), 81.1 (C-1C), 80.2 (C-3D), 79.8 (C-4D), 78.1 (C-4C), 75.9 (PhCH2), 74.8 (PhCH2), 74.5 (C-3C), 74.4 (C-2C), 73.3 (PhCH2), 71.8 (PhCH2), 70.4 (C-5C), 68.3 (C-5D), 67.8 (C-6D), 63.0 (C-2D), 25.4 (SCH2CH3), 17.8 (CCH3), 14.9 (SCH2CH3); ESIMS m/z: 868.3 [M + Na]+; anal. calcd for C49H55N3O8S (845.37): C, 69.56; H, 6.55; found: C, 69.33; H, 6.80.
4-Methoxyphenyl (2-azido-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl)-(1→3)-(2,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1→4)-(2,3-di-O-acetyl-6-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-acetyl-β-D-glucopyranoside (8): To a solution of compound 2 (1.2 g, 1.60 mmol) and compound 7 (1.6 g, 1.89 mmol) in anhydrous CH2Cl2 (8 mL) MS 4Å (2.0 g) was added and the reaction mixture was cooled to −10 °C. To the cooled reaction mixture NIS (500.0 mg, 2.22 mmol) and HClO4–SiO2 (20.0 mg) were added and it was allowed to stir for 1 h at same temperature. The reaction mixture was filtered through a bed of Celite® and washed with CH2Cl2 (100 mL). The organic layer was successively washed with 5% aq. Na2S2O3, NaHCO3 solution and water, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over silica gel using hexane–EtOAc (2:1) as eluant to give pure compound 8 (2.0 g, 82%). White solid; mp 66–68 °C; [α]D25 +18.2 (c 1.2, CHCl3); IR (KBr): 3428, 3032, 2932, 2108, 1753, 1508, 1455, 1368, 1230, 1044, 740, 698 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.35–7.05 (m, 30H, Ar-H), 6.91 (d, J = 9.0 Hz, 2H, Ar-H), 6.80 (J = 9.0 Hz, 2H, Ar-H), 5.34 (d, J = 4.5 Hz, 1H, H-1B), 5.29 (t, J = 10.0 Hz, 1H, H-3A), 5.27 (t, J = 9.0 Hz, 1H, H-3B), 5.04 (dd, J = 8.0 Hz each, 1H, H-2A), 4.97 (d, J = 3.5 Hz, 1H, H-1D), 4.95 (d, J = 8.0 Hz, 1H, H-1A), 4.89 (d, J = 2.0 Hz, 1H, H-1C), 4.85–4.80 (m, 2H, PhCH2), 4.78–4.75 (m, 3H, H-2B, PhCH2), 4.65 (d, J = 12.0 Hz, 1H, PhCH2), 4.60 (d, J = 12.0 Hz, 1H, PhCH2), 4.58–4.44 (m, 5H, PhCH2), 4.41 (dd, J = 12.5, 2.5 Hz, 1H, H-6aA), 4.35 (d, J = 12.0 Hz, 1H, PhCH2), 4.21 (dd, J = 12.5, 2.5 Hz, 1H, H-6bA), 4.05–4.01 (2 t, J = 9.5 Hz each, 2H, H-3D, H-4C), 3.98–3.92 (m, 2H, H-3c, H-5c), 3.90 (t, J = 10.0 Hz, 1H, H-4D), 3.81 (t, J = 8.5 Hz, 1H, H-4A), 3.77 (s, 3H, OCH3), 3.76–3.74 (m, 1H, H-4B), 3.72–3.71 (m, 1H, H-5B), 3.69–3.65 (m, 2H, H-5A, H-6aB), 3.63–3.61 (m, 1H, H-6aD), 3.58–3.55 (m, 3H, H-2C, H-5D, H-6bB), 3.53–3.51 (m, 1H, H-6bD), 3.43 (dd, J = 10.0, 3.5 Hz, 1H, H-2D), 2.04, 2.02, 2.01, 1.99, 1.95 (5 s, 15H, 5 COCH3), 1.25 (d, J = 6.2 Hz, 3H, CCH3); 13C NMR (150 MHz, CDCl3) δ 170.7, 170.2 (2C), 169.8, 169.7 (5 COCH3), 155.7–114.5 (Ar-C), 99.7 (C-1A), 98.8 (C-1C), 96.0 (C-1B), 93.9 (C-1D), 80.2 (C-3D), 79.4 (C-4D), 78.2 (C-5A), 77.2 (C-5B), 75.5 (PhCH2), 75.4 (C-3A), 75.3 (PhCH2), 74.8 (PhCH2), 74.3 (C-2B), 74.2 (C-4C), 73.7 (PhCH2), 73.3 (PhCH2), 72.6 (2C, C-5D, PhCH2), 72.2 (C-4A), 72.1 (C-2A), 71.3 (C-4B), 70.7 (2C, C-3C, C-5C), 70.4 (C-3B), 69.0 (C-2C), 67.9 (C-6B), 67.8 (C-6D), 63.3 (C-2D), 62.6 (C-6A), 55.6 (OCH3), 21.0, 20.9, 20.7, 20.6 (2C) (5 COCH3), 17.9 (CCH3); ESIMS m/z: 1554.6 [M + Na]+; anal. calcd for C83H93N3O25 (1531.61): C, 65.04; H, 6.12; found: C, 64.82; H, 6.36.
4-Methoxyphenyl (2-acetamido-2-deoxy-α-D-glucopyranosyl)-(1→3)-(α-L-rhamnopyranosyl)-(1→4)-(α-D-glucopyranosyl)-(1→4)-sodium β-D-glucopyranosid uronate (1): A solution of compound 8 (1.3 g, 0.85 mmol) in 0.1 M CH3ONa in CH3OH (25 mL) was allowed to stir at rt for 3 h and neutralized with Dowex 50W X8 (H+) resin. The reaction mixture was filtered and concentrated under reduced pressure. To a solution of the crude product in CH2Cl2 (25 mL) and H2O (4 mL) were sequentially added aq. NaBr (2 mL, 1 M), aq. TBAB (2.5 mL, 1 M), TEMPO (100.0 mg, 0.64 mmol), satd. NaHCO3 solution (10 mL) and 4% aq. NaOCl (15 mL) were added and the reaction mixture was allowed to stir at 0–5 °C for 2 h. The reaction mixture was neutralized with 1 N aq. HCl. To the reaction mixture tert-butanol (25 mL), 2-methylbut-2-ene (20 mL, 2 M solution in THF), aq. NaClO2 (1.5 g in 5 mL) and aq. NaH2PO4 (1.5 g in 5 mL) were added and the reaction mixture was stirred at room temperature for 3 h. The reaction mixture was diluted with satd. aq. NaH2PO4 and extracted with CH2Cl2 (150 mL). The organic layer was washed with water, dried (Na2SO4) and concentrated to dryness. The crude product was passed through a short pad of silica gel using EtOAc–toluene (2:1) as eluant. To a solution of the oxidized product (800.0 mg) in CH3OH–EtOAc (20 mL, 10:1 v/v) 20% Pd(OH)2/C (150.0 mg) was added and the reaction mixture was allowed to stir at room temperature under a positive pressure of hydrogen for 10 h. The reaction mixture was filtered through a bed of Celite® and evaporated to dryness. To a solution of the crude product in CH3OH (10 mL) acetic anhydride (2 mL) was added and the solution was kept at rt for 30 min. The solvents were removed under reduced pressure and the product was passed through a Sephadex® LH-20 column using CH3OH–H2O (60 mL, 4:1 v/v) as eluant to give pure compound 1 (450.0 mg, 64%). White powder; [α]D25 +14 (c 1.0, CH3OH); IR (KBr): 3428, 2937, 1621, 1366, 1152, 1087, 669 cm−1; 1H NMR (600 MHz, CD3OD) δ 7.04 (d, J = 9.0 Hz, 2H, Ar-H), 6.84 (d, J = 9.0 Hz, 2H, Ar-H), 5.20 (d, J = 3.6 Hz, 1H, H-1B), 4.93 (br s, 1H, H-1D), 4.89 (br s, 1H, H-1C), 4.82 (d, J = 7.8 Hz, 1H, H-1A), 4.03–3.98 (m, 2H, H-2A, H-5C), 3.97–3.95 (m, 2H, H-2C, H-2D), 3.88–3.86 (m, 1H, H-6aD), 3.83–3.75 (m, 5H, H-3A, H-3B, H-5D, H-6aB, H-6bD), 3.74 (s, 3H, OCH3), 3.73–3.71 (m, 1H, H-4A), 3.70–3.67 (m, 2H, H-2B, H-6bB), 3.64 (t, J = 9.6 Hz, 1H, H-3D), 3.56–3.50 (m, 4H, H-4B, H-4C, H-5A, H-5B), 3.49–3.44 (m, 2H, H-3C, H-4D), 2.02 (s, 3H, COCH3), 1.30 (d, J = 6.1Hz, 3H, CCH3); 13C NMR (150 MHz, CDCl3) δ 175.0 (COONa), 172.6 (NHCOCH3), 156.7–115.5 (Ar-C), 103.3 (C-1A), 102.8 (C-1B), 102.4 (C-1C), 96.7 (C-1D), 81.2 (C-3D), 79.5 (C-4B), 78.1 (C-3A), 77.7 (C-4A), 76.6 (C-5A), 74.6 (C-3C), 74.2 (C-4C), 73.8 (C-2B), 73.6 (C-2A), 73.0 (C-5D), 72.0 (C-5B), 71.9 (C-4D), 70.8 (C-5C), 69.3 (C-2C), 62.1 (C-6B), 62.0 (C-6D), 56.2 (OCH3), 55.3 (C-2D), 23.1 (NHCOCH3), 18.1 (CCH3); ESIMS m/z: 834.2 [M + H]+; anal. calcd for C33H48NNaO22 (833.26): C, 47.54; H, 5.80; found: C, 47.72; H, 6.07.
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra of compounds 2, 6, 7, 8 and 1. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
von Seidlein, L.; Kim, D. R.; Ali, M.; Lee, H.; Wang, X.; Thiem, V. D.; Canh, D. G.; Chaicumpa, W.; Agtini, M. D.; Hossain, A.; Bhutta, Z. A.; Mason, C.; Sethabutr, O.; Talukder, K.; Nair, G. B.; Deen, J. L.; Kotloff, K.; Clemens, J. PLoS Med. 2006, 1556–1569.
Return to citation in text: [1] -
Wachsmuth, K.; Morris, G. K. In Shigella, Foodborne Bacterial Pathogens; Doyle, M. P., Ed.; Marcel Dekker Inc: New York, 1989; pp 447–462.
Return to citation in text: [1] -
Gupta, A.; Polyak, C. S.; Bishop, R. D.; Sobel, J.; Mintz, E. D. Clin. Infect. Dis. 2004, 38, 1372–1377. doi:10.1086/386326
Return to citation in text: [1] -
Liu, B.; Knirel, Y. A.; Feng, L.; Perepelov, A. V.; Senchenkova, S. N.; Wang, Q.; Reeves, P. R.; Wang, L. FEMS Microbiol. Rev. 2008, 32, 627–653. doi:10.1111/j.1574-6976.2008.00114.x
Return to citation in text: [1] -
Wathen-Grady, H. G.; Davis, B. R.; Morris, G. K. J. Clin. Microbiol. 1985, 21, 129–132.
Return to citation in text: [1] -
Reeves, P. R.; Wang, L. Curr. Top. Microbiol. Immunol. 2002, 264, 109–135.
Return to citation in text: [1] -
Knirel, Y. A.; Kochetkov, N. K. Biochemistry (Moscow) 1994, 59, 1325–1383.
Return to citation in text: [1] -
L’vov, V. L.; Musina, L. I.; Shashkov, A. S.; Ermakov, G. P.; Dmitriev, B. A. Bioorg. Khim. 1987, 13, 1245–1255.
Return to citation in text: [1] -
Kosek, M.; Yori, P. P.; Olortegui, M. P. Curr. Opin. Infect. Dis. 2010, 23, 475–480. doi:10.1097/QCO.0b013e32833da204
Return to citation in text: [1] -
Bhan, M. K.; Raj, P.; Srivastava, R.; Bhandari, N.; Kumar, R. Indian Pediatr. 1988, 25, 804–805.
Return to citation in text: [1] -
Klee, S. R.; Tzschaschel, B. D.; Singh, M.; Falt, I.; Lindberg, A. A.; Timmis, K. N.; Guzman, C. A. Microb. Pathog. 1997, 22, 363–376. doi:10.1006/mpat.1996.0127
Return to citation in text: [1] -
Kweon, M.-N. Curr. Opin. Infect. Dis. 2008, 21, 313–318. doi:10.1097/QCO.0b013e3282f88b92
Return to citation in text: [1] -
Niyogi, S. K. J. Microbiol. 2005, 43, 133–143.
Return to citation in text: [1] -
Oaks, E. V.; Turbyfill, K. R. Vaccine 2006, 24, 2290–2301. doi:10.1016/j.vaccine.2005.11.040
Return to citation in text: [1] -
Pozsgay, V.; Kubler-Kielb, J. ACS Symposium Ser. 2007, 960, 238–252. doi:10.1021/bk-2007-0960.ch014
Return to citation in text: [1] -
Phalipon, A.; Costachel, C.; Grandjean, C.; Thuizat, A.; Guerreiro, C.; Tanguy, M.; Nato, F.; Vulliez-Le Normand, B.; Belot, F.; Wright, K.; Marcel-Peyre, V.; Sansonetti, P. J.; Mulard, L. A. J. Immunol. 2006, 176, 1686–1694.
Return to citation in text: [1] -
Pozsgay, V.; Coxon, B.; Glaudemans, C. P. J.; Schneerson, R.; Robbins, J. B. Synlett 2003, 743–767. doi:10.1055/s-2003-38724
Return to citation in text: [1] -
Pozsgay, V. Angew. Chem., Int. Ed. 1998, 37, 138–142. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<138::AID-ANIE138>3.0.CO;2-T
Return to citation in text: [1] -
Carlin, N. I.; Bundle, D. R.; Lindberg, A. A. J. Immunol. 1987, 138, 4419–4427.
Return to citation in text: [1] -
Santra, A.; Guchhait, G.; Misra, A. K. Green Chem. 2011, 13, 1345–1351. doi:10.1039/c1gc15122c
Return to citation in text: [1] [2] -
Panchadhayee, R.; Misra, A. K. Synlett 2010, 1193–1196. doi:10.1055/s-0029-1219798
Return to citation in text: [1] [2] -
Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007
Return to citation in text: [1] [2] -
Ludek, O. R.; Gu, W.; Gildersleeve, J. C. Carbohydr. Res. 2010, 345, 2074–2078. doi:10.1016/j.carres.2010.07.030
Return to citation in text: [1] [2] -
Du, Y.; Wei, G.; Cheng, S.; Hua, Y.; Linhardt, R. J. Tetrahedron Lett. 2006, 47, 307–310. doi:10.1016/j.tetlet.2005.11.025
Return to citation in text: [1] [2] -
Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913
Return to citation in text: [1] [2] -
Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119
Return to citation in text: [1] [2] -
Huang, L.; Teumelsan, N.; Huang, X. Chem.–Eur. J. 2006, 12, 5246–5252. doi:10.1002/chem.200600290
Return to citation in text: [1] [2] -
Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 111–119. doi:10.1007/s10719-007-9061-0
Return to citation in text: [1] [2] -
Panchadhayee, R.; Misra, A. K. Tetrahedron: Asymmetry 2009, 20, 1550–1555. doi:10.1016/j.tetasy.2009.05.026
Return to citation in text: [1] [2] -
Attolino, E.; Rising, T. W. D. F.; Heidecke, C. D.; Fairbanks, A. J. Tetrahedron: Asymmetry 2007, 18, 1721–1734. doi:10.1016/j.tetasy.2007.06.026
Return to citation in text: [1] -
Werz, D. B.; Seeberger, P. H. Angew. Chem., Int. Ed. 2005, 44, 6315–6318. doi:10.1002/anie.200502615
Return to citation in text: [1] -
Marmuse, L.; Nepogodiev, S. A.; Field, R. A. Tetrahedron: Asymmetry 2005, 16, 477–485. doi:10.1016/j.tetasy.2004.11.047
Return to citation in text: [1] -
Grundler, G.; Schmidt, R. R. Liebigs Ann. Chem. 1984, 1826–1847. doi:10.1002/jlac.198419841108
Return to citation in text: [1] -
Sajtos, F.; Hajko, J.; Kover, K. E.; Liptak, A. Carbohydr. Res. 2001, 334, 253–259. doi:10.1016/S0008-6215(01)00196-3
Return to citation in text: [1] -
Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1986, 25, 212–235. doi:10.1002/anie.198602121
Return to citation in text: [1] -
Bock, K.; Pedersen, C. Acta Chem. Scand., Ser. B 1975, 29, 258–264. doi:10.3891/acta.chem.scand.29b-0181
Return to citation in text: [1] -
Duus, J. O.; Gotfredsen, C. H.; Bock, K. Chem. Rev. 2000, 100, 4589–4614. doi:10.1021/cr990302n
Return to citation in text: [1] -
Crich, D.; Li, H. J. Org. Chem. 2002, 67, 4640–4646. doi:10.1021/jo0108818
Return to citation in text: [1] -
Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f
Return to citation in text: [1]
| 1. | von Seidlein, L.; Kim, D. R.; Ali, M.; Lee, H.; Wang, X.; Thiem, V. D.; Canh, D. G.; Chaicumpa, W.; Agtini, M. D.; Hossain, A.; Bhutta, Z. A.; Mason, C.; Sethabutr, O.; Talukder, K.; Nair, G. B.; Deen, J. L.; Kotloff, K.; Clemens, J. PLoS Med. 2006, 1556–1569. |
| 2. | Wachsmuth, K.; Morris, G. K. In Shigella, Foodborne Bacterial Pathogens; Doyle, M. P., Ed.; Marcel Dekker Inc: New York, 1989; pp 447–462. |
| 3. | Gupta, A.; Polyak, C. S.; Bishop, R. D.; Sobel, J.; Mintz, E. D. Clin. Infect. Dis. 2004, 38, 1372–1377. doi:10.1086/386326 |
| 8. | L’vov, V. L.; Musina, L. I.; Shashkov, A. S.; Ermakov, G. P.; Dmitriev, B. A. Bioorg. Khim. 1987, 13, 1245–1255. |
| 32. | Marmuse, L.; Nepogodiev, S. A.; Field, R. A. Tetrahedron: Asymmetry 2005, 16, 477–485. doi:10.1016/j.tetasy.2004.11.047 |
| 6. | Reeves, P. R.; Wang, L. Curr. Top. Microbiol. Immunol. 2002, 264, 109–135. |
| 7. | Knirel, Y. A.; Kochetkov, N. K. Biochemistry (Moscow) 1994, 59, 1325–1383. |
| 20. | Santra, A.; Guchhait, G.; Misra, A. K. Green Chem. 2011, 13, 1345–1351. doi:10.1039/c1gc15122c |
| 5. | Wathen-Grady, H. G.; Davis, B. R.; Morris, G. K. J. Clin. Microbiol. 1985, 21, 129–132. |
| 27. | Huang, L.; Teumelsan, N.; Huang, X. Chem.–Eur. J. 2006, 12, 5246–5252. doi:10.1002/chem.200600290 |
| 28. | Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 111–119. doi:10.1007/s10719-007-9061-0 |
| 29. | Panchadhayee, R.; Misra, A. K. Tetrahedron: Asymmetry 2009, 20, 1550–1555. doi:10.1016/j.tetasy.2009.05.026 |
| 4. | Liu, B.; Knirel, Y. A.; Feng, L.; Perepelov, A. V.; Senchenkova, S. N.; Wang, Q.; Reeves, P. R.; Wang, L. FEMS Microbiol. Rev. 2008, 32, 627–653. doi:10.1111/j.1574-6976.2008.00114.x |
| 30. | Attolino, E.; Rising, T. W. D. F.; Heidecke, C. D.; Fairbanks, A. J. Tetrahedron: Asymmetry 2007, 18, 1721–1734. doi:10.1016/j.tetasy.2007.06.026 |
| 31. | Werz, D. B.; Seeberger, P. H. Angew. Chem., Int. Ed. 2005, 44, 6315–6318. doi:10.1002/anie.200502615 |
| 20. | Santra, A.; Guchhait, G.; Misra, A. K. Green Chem. 2011, 13, 1345–1351. doi:10.1039/c1gc15122c |
| 22. | Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007 |
| 15. | Pozsgay, V.; Kubler-Kielb, J. ACS Symposium Ser. 2007, 960, 238–252. doi:10.1021/bk-2007-0960.ch014 |
| 16. | Phalipon, A.; Costachel, C.; Grandjean, C.; Thuizat, A.; Guerreiro, C.; Tanguy, M.; Nato, F.; Vulliez-Le Normand, B.; Belot, F.; Wright, K.; Marcel-Peyre, V.; Sansonetti, P. J.; Mulard, L. A. J. Immunol. 2006, 176, 1686–1694. |
| 17. | Pozsgay, V.; Coxon, B.; Glaudemans, C. P. J.; Schneerson, R.; Robbins, J. B. Synlett 2003, 743–767. doi:10.1055/s-2003-38724 |
| 18. | Pozsgay, V. Angew. Chem., Int. Ed. 1998, 37, 138–142. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<138::AID-ANIE138>3.0.CO;2-T |
| 19. | Carlin, N. I.; Bundle, D. R.; Lindberg, A. A. J. Immunol. 1987, 138, 4419–4427. |
| 23. | Ludek, O. R.; Gu, W.; Gildersleeve, J. C. Carbohydr. Res. 2010, 345, 2074–2078. doi:10.1016/j.carres.2010.07.030 |
| 24. | Du, Y.; Wei, G.; Cheng, S.; Hua, Y.; Linhardt, R. J. Tetrahedron Lett. 2006, 47, 307–310. doi:10.1016/j.tetlet.2005.11.025 |
| 25. | Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913 |
| 26. | Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 11. | Klee, S. R.; Tzschaschel, B. D.; Singh, M.; Falt, I.; Lindberg, A. A.; Timmis, K. N.; Guzman, C. A. Microb. Pathog. 1997, 22, 363–376. doi:10.1006/mpat.1996.0127 |
| 12. | Kweon, M.-N. Curr. Opin. Infect. Dis. 2008, 21, 313–318. doi:10.1097/QCO.0b013e3282f88b92 |
| 13. | Niyogi, S. K. J. Microbiol. 2005, 43, 133–143. |
| 14. | Oaks, E. V.; Turbyfill, K. R. Vaccine 2006, 24, 2290–2301. doi:10.1016/j.vaccine.2005.11.040 |
| 9. | Kosek, M.; Yori, P. P.; Olortegui, M. P. Curr. Opin. Infect. Dis. 2010, 23, 475–480. doi:10.1097/QCO.0b013e32833da204 |
| 10. | Bhan, M. K.; Raj, P.; Srivastava, R.; Bhandari, N.; Kumar, R. Indian Pediatr. 1988, 25, 804–805. |
| 21. | Panchadhayee, R.; Misra, A. K. Synlett 2010, 1193–1196. doi:10.1055/s-0029-1219798 |
| 34. | Sajtos, F.; Hajko, J.; Kover, K. E.; Liptak, A. Carbohydr. Res. 2001, 334, 253–259. doi:10.1016/S0008-6215(01)00196-3 |
| 21. | Panchadhayee, R.; Misra, A. K. Synlett 2010, 1193–1196. doi:10.1055/s-0029-1219798 |
| 33. | Grundler, G.; Schmidt, R. R. Liebigs Ann. Chem. 1984, 1826–1847. doi:10.1002/jlac.198419841108 |
| 39. | Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f |
| 36. | Bock, K.; Pedersen, C. Acta Chem. Scand., Ser. B 1975, 29, 258–264. doi:10.3891/acta.chem.scand.29b-0181 |
| 37. | Duus, J. O.; Gotfredsen, C. H.; Bock, K. Chem. Rev. 2000, 100, 4589–4614. doi:10.1021/cr990302n |
| 38. | Crich, D.; Li, H. J. Org. Chem. 2002, 67, 4640–4646. doi:10.1021/jo0108818 |
| 27. | Huang, L.; Teumelsan, N.; Huang, X. Chem.–Eur. J. 2006, 12, 5246–5252. doi:10.1002/chem.200600290 |
| 28. | Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 111–119. doi:10.1007/s10719-007-9061-0 |
| 29. | Panchadhayee, R.; Misra, A. K. Tetrahedron: Asymmetry 2009, 20, 1550–1555. doi:10.1016/j.tetasy.2009.05.026 |
| 22. | Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007 |
| 25. | Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913 |
| 26. | Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 35. | Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1986, 25, 212–235. doi:10.1002/anie.198602121 |
| 23. | Ludek, O. R.; Gu, W.; Gildersleeve, J. C. Carbohydr. Res. 2010, 345, 2074–2078. doi:10.1016/j.carres.2010.07.030 |
| 24. | Du, Y.; Wei, G.; Cheng, S.; Hua, Y.; Linhardt, R. J. Tetrahedron Lett. 2006, 47, 307–310. doi:10.1016/j.tetlet.2005.11.025 |
© 2011 Santra and Misra; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)