Abstract
Fluorinated and nonfluorinated phosphonates are employed as precatalysts in lithium phosphonate catalyzed cross benzoin couplings. Surprisingly, a decreased catalytic activity for the fluorinated precatalysts compared to the nonfluorinated systems is observed. Furthermore, the ring size of six, seven and nine membered ring catalysts appears not to be crucial for their catalytic activity.

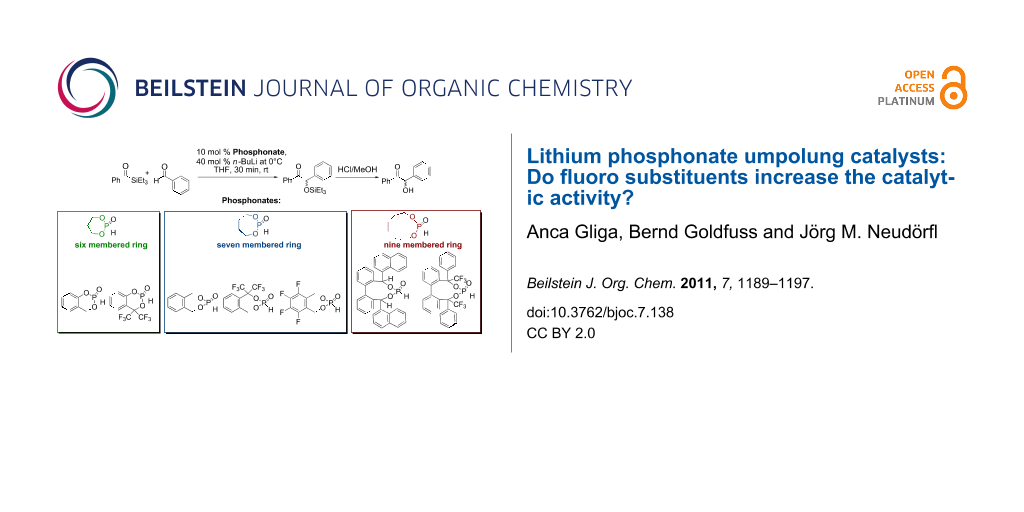
Graphical Abstract
Introduction
Since the discovery of the cyanide catalyzed benzoin reaction by Liebig and Wöhler in 1832 [1], acyloin-type reactions evolved as powerful tools for couplings of acylanion equivalents with carbon electrophiles. In addition to cyanide [2-5] and nucleophilic carbene catalysts (e.g. thiazolium salts) [6-17], lithium phosphonates were found to catalyze cross acyloin type couplings of acylsilanes with aldehydes [18]. The catalytic cycle proposed by Johnson et al. [18] (Scheme 1) suggests that a potential metallophosphonate catalyst must act as a nucleophile, an anion (d1-synthon) stabilization group, and as well as a leaving group (nucleofuge). Comparative computational assessments of carbanionic d1-species, which have been proposed as crucial intermediates according to the Lapworth and Breslow mechanisms, show comparable activities for lithium phosphonate and cyanide [19,20].
![[1860-5397-7-138-i1]](/bjoc/content/inline/1860-5397-7-138-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed catalytic cycle of the lithium phosphonate catalyzed cross benzoin coupling [18].
Scheme 1: Proposed catalytic cycle of the lithium phosphonate catalyzed cross benzoin coupling [18].
Recently, we introduced fenchol based phosphonates as precatalysts, which are similarly accessible as fencholate metal catalysts [21-25], in the benzoin coupling (Scheme 2) [26]. A strong increase of the catalytic activity was observed for a benzylic fencholate, when the benzylic positions were occupied by CF3-groups (92% versus 19% yield, Scheme 2) [26]. This increased reactivity is thought to arise from a favored formation of the carbanionic d1-synthon intermediate, due to the electron withdrawing effect of the CF3 groups. A comparison of fluorinated and nonfluorinated TADDOL phosphonates (which were used by Johnson's group) as precatalysts in benzoin coupling does not show any difference in reactivity (Scheme 2). In contrast the enantioselectivity is clearly higher with the fluorinated TADDOL precatalyst (Scheme 2).
![[1860-5397-7-138-i2]](/bjoc/content/inline/1860-5397-7-138-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Phosphonate as precatalysts in benzoin coupling [18,26].
Scheme 2: Phosphonate as precatalysts in benzoin coupling [18,26].
Here, we analyze the effect of fluoro substituents on the catalytic activity by using different fluorinated and nonfluorinated phosphonates as precatalysts in the benzoin coupling.
Results and Discussion
As precursors for six, seven and nine membered ring phosphonates, diols 1–4, and 6–8 were synthesized (Scheme 3). The synthesis of diol 1 was conducted by an ortho lithiation of 1,1,1,3,3,3-hexafluoro-2-phenylpropan-2-ol and subsequent addition of the in situ generated carbanion to formaldehyde. For comparison, a nonfluorinated diol precursor 2 was synthesized. Diol 3 was used as the precursor to investigate the influence of aromatic fluoro substituents. Six ring phosphonates were realized with diol 4 [27] and the analogous nonfluorinated 2-(hydroxymethyl)phenol (5) as precursor.
![[1860-5397-7-138-i3]](/bjoc/content/inline/1860-5397-7-138-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of diols 1–4, 6–7; diol 5 is commercially available.
Scheme 3: Synthesis of diols 1–4, 6–7; diol 5 is commercially available.
Biphenyl-based fluorinated and nonfluorinated systems (6 [28-30], 7 and 8, Scheme 3) were chosen as precursors for the synthesis of nine ring phosphonates. The synthesis of these diols was realized by a double ortho lithiation of biphenyl and subsequent addition to the corresponding carbonyl compound. By this procedure, two asymmetric carbon centres and a chiral axis, which is fixed by intramolecular hydrogen bonds (6: Intramolecular O1–O2 distance 2.83 Å, 7: Intramolecular O1–O2 distance 2.81 Å, Figure 1 and Figure 2, respectively), are generated. Chiral HPLC and X-ray analyses revealed one pair of enantiomers for diol 6 and 7 (HPLC (Daicel-OD-H, 90:10 n-hexane/isopropanol; flow 0.5 mL/min): 6: tR1 = 10.4 min; tR2 = 13.2 min (racemate); 7: tR = 22.1 min; X-ray structures shown in Figure 1 and Figure 2, respectively) and an additional meso product for diol 8 (HPLC: tR2 = 22.4 min; tR3 = 30.6 min (racemate); tR1 = 9.1 min). A dimer associated by a hydrogen bond is apparent for the enantiomeric pair (6: intermolecular O1–O2 distance 2.81 Å, 7: intermolecular O1–O2 distance 2.80 Å, Figure 1 and Figure 2, respectively). The favored formed enantiomeric pair has the same configuration (R,R or S,S) for both benzylic carbon centres, which defines the conformation of the biphenyl axis (M(inus) for S,S and P(lus) for R,R).
![[1860-5397-7-138-1]](/bjoc/content/figures/1860-5397-7-138-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of 6. (M)-(S,S) and (P)-(R,R) pair of enantiomers; intermolecular O1–O2 distance 2.81 Å; intramolecular O1–O2 distance 2.83 Å. Ellipsoids correspond to 50% probability levels. Hydrogen atoms are omitted for clarity.
Figure 1: X-ray crystal structure of 6. (M)-(S,S) and (P)-(R,R) pair of enantiomers; intermolecular O1–O2 dis...
![[1860-5397-7-138-2]](/bjoc/content/figures/1860-5397-7-138-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of 7. (M)-(S,S) and (P)-(R,R) pair of enantiomers; intermolecular O1–O2 distance 2.80 Å; intramolecular O1–O2 distance 2.81 Å. Ellipsoids correspond to 50% probability levels. Hydrogen atoms are omitted for clarity.
Figure 2: X-ray crystal structure of 7. (M)-(S,S) and (P)-(R,R) pair of enantiomers; intermolecular O1–O2 dis...
The conformational stability of the biphenyl axis can be demonstrated by the energy difference of the optimized structures (B3LYP 6-31G*) (Table 1) (between (M)-(S,S); (P)-(R,R) and (P)-(S,S); (M)-(R,R) for 6 Erel = 3.6 kcal/mol; for 7 Erel = 4.3 kcal/mol). The alternative diastereomers, with different configurations at the benzylic carbon (M)-(R,S); (P)-(R,S), are energetically disfavored (Table 1). For these diastereomers two possible directions of the hydrogen bond were considered, that is (M)-(R,S); (P)-(R,S) with the hydrogen bond from S→R giving Erel = 1.4 kcal/mol and (M)-(R,S); (P)-(R,S) with the hydrogen bond from R→S giving Erel = 1.8 kcal/mol for 6; equivalently Erel = 4.7 kcal/mol and Erel = 4.4 kcal/mol in the respective cases for 7. Similar biphenyl conformation stabilities were found for 1,1′-biphenyl-2,2′-bisterpenols (terpenol moiety: (−)-Fenchol, (−)-menthol, (−)-verbenol und (−)-carvol) [31,32]. The energy differences of the terpene-based conformers are between 5.1 and 5.8 kcal/mol [32,33] (B3LYP/6-31++G**:AM1).
Table 1: Optimized structures of diols 6 and 7.
| diol 6 | ||||
|---|---|---|---|---|
| diastereomer | (M)-(S,S); (P)-(R,R) | (P)-(S,S); (M)-(R,R) |
(M)-(R,S);
(P)-(R,S) R→Sa |
(M)-(R,S);
(P)-(R,S) S→Ra |
| Erel [kcal/mol]b | 0 | +3.6 | +1.8 | +1.4 |
| diol 7 | ||||
| diastereomer | (M)-(S,S); (P)-(R,R) | (P)-(S,S); (M)-(R,R) |
(M)-(R,S);
(P)-(R,S) R→Sa |
M)-(R,S);
(P)-(R,S) S→Ra |
| Erel [kcal/mol]b | 0 | +4.3 | +4.4 | +4.7 |
aDirection of hydrogen bond; bB3LYP 6-31G*.
The conversion of diols 1–5, 7, and 8 to the desired phosphonates can be achieved by twofold addition to phosphorus trichloride and subsequent hydrolysis. Diol 6 could not be converted under the employed conditions (Scheme 4).
![[1860-5397-7-138-i4]](/bjoc/content/inline/1860-5397-7-138-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of phosphonates 9–15.
Scheme 4: Synthesis of phosphonates 9–15.
The strong inductive effect of the fluoro substituents is clearly visible from the 1J(P–H) coupling constants, which are significantly increased compared to the nonfluoro-substituted phosphonates (Table 2). In general, 1J(P–H) coupling constants increase with the electronegativity of the substituents [33]. The influence of electronegativity results from the change in s-character, given that the Fermi-contact is the dominant coupling mechanism [33]. According to Bent’s rule [34] electron withdrawing substituents require more p-character in the bonding orbitals, which leads to an increased s-character in the bonding P–H orbital. The smallest influence on the coupling constant is apparent for phosphonate 11, in which the fluoro substituents are not in close proximity to the phosphorous atom (five bonds distance). The largest 1J(P–H) coupling constant was detected for phosphonate 13. Thereby an additional electron withdrawing effect of the phenoxy group causes the further increase in the 1J(P–H) coupling constant. All synthesized phosphonates were identified by 31P NMR, especially characteristic are the phosphorus–hydrogen and phosphorus–fluoro couplings.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-138-i6.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-138-i7.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-138-i8.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-138-i9.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-138-i10.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-138-i11.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-138-i12.svg?max-width=637&scale=1.0)
For phosphonates 14 and 15 (Figure 3) a doublet splitting caused by the 1J(P–H) coupling ~700 Hz was observed in the 31P NMR spectra. The protons in the benzylic position (phosphonate 14) effect a 3J(P–H) coupling (12.5 Hz) and the CF3 groups in this position (phosphonate 15) a 4J(P–F) coupling (14.3 Hz) (Figure 3).
![[1860-5397-7-138-3]](/bjoc/content/figures/1860-5397-7-138-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 31P NMR of phosphonate 14 (a) and 15 (b).
Figure 3: 31P NMR of phosphonate 14 (a) and 15 (b).
A crystal structure was obtained for phosphonate 15, which shows the (M)-(R,S) diastereomers (Figure 4).
![[1860-5397-7-138-4]](/bjoc/content/figures/1860-5397-7-138-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structure of 15. (M)-(R,S) diastereomer; ellipsoids correspond to 50% probability levels. Hydrogen atoms are omitted for clarity.
Figure 4: X-ray crystal structure of 15. (M)-(R,S) diastereomer; ellipsoids correspond to 50% probability lev...
The lithium phosphonate catalyzed benzoin reaction (Scheme 5) with phosphonates 9–15 as precatalysts led to the benzoin product in low to moderate yields (5–44%) (Figure 5). The supposed increase in catalytic activity, which was observed for fenchol-based phosphonate (Scheme 2) [21] with fluoro-substituted phosphonates as precatalysts, could not be confirmed. The highest yield was achieved with phosphonate 14 as precatalyst (44%). Contrary to expectations this yield is twice as high as the yield achieved with phosphonate 15. The reduction of the nucleophilic character of the phosphorus nucleophile in the first step of the catalytic cycle, and of the d1-synthon in the third step of the catalytic cycle (Scheme 1), could explain these results. The increased 1J(P–H) coupling constants for the CF3 substituted phosphonates (10, 13, 15, Table 2) suggest an increase in s-character at the phosphorus atom, which confirms a reduction in nucleophilic character. In contrast to the fenchol-based phosphonates [26] the best result was achieved by a nine membered ring phosphonate instead of a seven membered ring phosphonate. It can be concluded that the ring size is not of basic importance to the catalytic activity.
![[1860-5397-7-138-i5]](/bjoc/content/inline/1860-5397-7-138-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Lithium phosphonate catalysts in cross benzoin coupling.
Scheme 5: Lithium phosphonate catalysts in cross benzoin coupling.
![[1860-5397-7-138-5]](/bjoc/content/figures/1860-5397-7-138-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Phosphonate precatalysts 9–15 in cross benzoin coupling.
Figure 5: Phosphonate precatalysts 9–15 in cross benzoin coupling.
Conclusion
Three types of cyclic fluorinated and nonfluorinated phosphonates were synthesized and used as precatalysts in cross benzoin couplings with yields ranging from 5 to 44%. The inductive effect of CF3 substituents in benzylic position of phosphonates 10, 13 and 15 gives rise to increased 1J(P–H) couplings in P–H precatalysts and hence points to increased s-character at the phosphorus lone pair in the active anionic catalysts [33]. A rise of catalytic activity due to the inductive effect of CF3 substituents, as was observed before for a fenchol-based phosphonate (Scheme 2) [26], was not realized with the phosphonates employed herein. Instead a reduction of catalytic activity was apparent with fluorinated phosphonates compared to the nonfluorinated phosphonates. This can be explained by a weaker nucleophilic character of the phosphorus nucleophile, as a consequence of the increased s-character. Comparisons of phosphonates with different ring sizes show that the nine ring phosphonates result in higher yields than do the six and seven ring phosphonates.
Supporting Information
Experimental procedures, characterization data for compounds 1–4, 6–15, crystallographic data for compounds 6, 7, 15, computational details for compounds 6, 7 and general procedure for phosphonates as precatalysts in cross benzoin coupling.
| Supporting Information File 1: Experimental procedure and characterization data. | ||
| Format: PDF | Size: 135.3 KB | Download |
Acknowledgements
We are grateful to the Fonds der Chemischen Industrie for financial support. We especially thank the Deutsche Forschungsgemeinschaft (DFG) for funding (GO-930/9-1) as well as the Bayer AG, the BASF AG, the Wacker AG, the Evonic AG, the Raschig GmbH, the Symrise GmbH, the Solvay GmbH and the OMG group for generous support.
References
-
Wöhler, F.; von Liebig, J. Ann. Pharm. 1832, 3, 249–282. doi:10.1002/jlac.18320030302
Return to citation in text: [1] -
Linghu, X.; Johnson, J. S. Angew. Chem. 2003, 115, 2638–2640. doi:10.1002/ange.200250554
Return to citation in text: [1] -
Tarr, J. C.; Johnson, J. S. Org. Lett. 2009, 11, 3870–3873. doi:10.1021/ol901314w
Return to citation in text: [1] -
Bausch, C. C.; Johnson, J. S. Adv. Synth. Catal. 2005, 347, 1207–1211. doi:10.1002/adsc.200505097
Return to citation in text: [1] -
Demir, A. S.; Reis, Ö.; İğdir, A. Ç.; Esiringü, İ.; Eymur, S. J. Org. Chem. 2005, 70, 10584–10587. doi:10.1021/jo051811u
Return to citation in text: [1] -
Ugai, T.; Tanaka, S.; Dokawa, S. J. Pharm. Soc. Jpn. 1943, 63, 296–300.
Return to citation in text: [1] -
Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719–3726. doi:10.1021/ja01547a064
Return to citation in text: [1] -
Sheehan, J. C.; Hunneman, D. H. J. Am. Chem. Soc. 1966, 88, 3666–3667. doi:10.1021/ja00967a049
Return to citation in text: [1] -
Sheehan, J. C.; Hara, T. J. Org. Chem. 1974, 39, 1196–1199. doi:10.1021/jo00923a006
Return to citation in text: [1] -
Tagaki, W.; Tamura, Y.; Yano, Y. Bull. Chem. Soc. Jpn. 1980, 53, 478–480. doi:10.1246/bcsj.53.478
Return to citation in text: [1] -
Enders, D.; Breuer, K.; Teles, J. H. Helv. Chim. Acta 1996, 79, 1217–1221. doi:10.1002/hlca.19960790427
Return to citation in text: [1] -
Knight, R. L.; Leeper, F. J. J. Chem. Soc., Perkin Trans. 1 1998, 1891–1894. doi:10.1039/A803635G
Return to citation in text: [1] -
Enders, D.; Kallfass, U. Angew. Chem., Int. Ed. 2002, 41, 1743–1745. doi:10.1002/1521-3773(20020517)41:10<1743::AID-ANIE1743>3.0.CO;2-Q
Return to citation in text: [1] -
Enders, D.; Niemeier, O. Angew. Chem., Int. Ed. 2006, 118, 1491–1495. doi:10.1002/anie.200503885
Return to citation in text: [1] -
Takikawa, H.; Hachisu, Y.; Bode, J. W.; Suzuki, K. Angew. Chem., Int. Ed. 2006, 118, 3572–3574. doi:10.1002/anie.200600268
Return to citation in text: [1] -
Enders, D.; Henseler, A. Adv. Synth. Catal. 2009, 351, 1749–1752. doi:10.1002/adsc.200900247
Return to citation in text: [1] -
Enders, D.; Grossmann, A.; Fronert, J.; Raabe, G. Chem. Commun. 2010, 46, 6282–6284. doi:10.1039/c0cc02013c
Return to citation in text: [1] -
Linghu, X.; Potnick, J. R.; Johnson, J. S. J. Am. Chem. Soc. 2004, 126, 3070–3071. doi:10.1021/ja0496468
Return to citation in text: [1] [2] [3] [4] -
Goldfuss, B.; Schumacher, M. J. Mol. Model. 2006, 12, 591–595. doi:10.1007/s00894-005-0036-4
Return to citation in text: [1] -
Schumacher, M.; Goldfuss, B. Tetrahedron 2008, 64, 1648–1653. doi:10.1016/j.tet.2007.12.019
Return to citation in text: [1] -
Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211–5218. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S
Return to citation in text: [1] [2] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785–1792. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273
Return to citation in text: [1] -
Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7
Return to citation in text: [1] -
Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 2, 256–263. doi:10.1002/ejoc.201001295
Return to citation in text: [1] [2] [3] [4] [5] -
Nolan, B. G.; Tsujioka, S.; Strauss, S. H. J. Fluorine Chem. 2002, 118, 103–106. doi:10.1016/S0022-1139(02)00203-8
Return to citation in text: [1] -
Bergmann, E. D.; Pelchowicz, Z. J. Org. Chem. 1954, 19, 1387–1390. doi:10.1021/jo01373a024
Return to citation in text: [1] -
Weitzberg, M.; Abu-Shakra, E.; Azeb, A.; Aizenshtat, Z.; Blum, J. J. Org. Chem. 1987, 52, 529–536. doi:10.1021/jo00380a010
Return to citation in text: [1] -
Tsantrizos, Y. S.; Folkins, P. L.; Britten, J. F.; Harpp, D. N.; Ogilvie, K. K. Can. J. Chem. 1992, 70, 158–164. doi:10.1139/v92-026
Return to citation in text: [1] -
Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7
Return to citation in text: [1] -
Alpagut, Y.; Goldfuss, B.; Neudörfel, J.-M. Beilstein J. Org. Chem. 2008, 4, No. 25. doi:10.3762/bjoc.4.25
Return to citation in text: [1] [2] -
Berger, S.; Braun, S.; Kalinowski, H.-O. NMR-Spektroskopie von Nichtmetallen Band 3. Thieme: Stuttgart, Germany, 1993; pp 121–122.
Return to citation in text: [1] [2] [3] [4] -
Bent, H. A. Chem. Rev. 1961, 61, 275–311. doi:10.1021/cr60211a005
Return to citation in text: [1]
| 21. | Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211–5218. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S |
| 33. | Berger, S.; Braun, S.; Kalinowski, H.-O. NMR-Spektroskopie von Nichtmetallen Band 3. Thieme: Stuttgart, Germany, 1993; pp 121–122. |
| 1. | Wöhler, F.; von Liebig, J. Ann. Pharm. 1832, 3, 249–282. doi:10.1002/jlac.18320030302 |
| 18. | Linghu, X.; Potnick, J. R.; Johnson, J. S. J. Am. Chem. Soc. 2004, 126, 3070–3071. doi:10.1021/ja0496468 |
| 32. | Alpagut, Y.; Goldfuss, B.; Neudörfel, J.-M. Beilstein J. Org. Chem. 2008, 4, No. 25. doi:10.3762/bjoc.4.25 |
| 33. | Berger, S.; Braun, S.; Kalinowski, H.-O. NMR-Spektroskopie von Nichtmetallen Band 3. Thieme: Stuttgart, Germany, 1993; pp 121–122. |
| 18. | Linghu, X.; Potnick, J. R.; Johnson, J. S. J. Am. Chem. Soc. 2004, 126, 3070–3071. doi:10.1021/ja0496468 |
| 33. | Berger, S.; Braun, S.; Kalinowski, H.-O. NMR-Spektroskopie von Nichtmetallen Band 3. Thieme: Stuttgart, Germany, 1993; pp 121–122. |
| 6. | Ugai, T.; Tanaka, S.; Dokawa, S. J. Pharm. Soc. Jpn. 1943, 63, 296–300. |
| 7. | Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719–3726. doi:10.1021/ja01547a064 |
| 8. | Sheehan, J. C.; Hunneman, D. H. J. Am. Chem. Soc. 1966, 88, 3666–3667. doi:10.1021/ja00967a049 |
| 9. | Sheehan, J. C.; Hara, T. J. Org. Chem. 1974, 39, 1196–1199. doi:10.1021/jo00923a006 |
| 10. | Tagaki, W.; Tamura, Y.; Yano, Y. Bull. Chem. Soc. Jpn. 1980, 53, 478–480. doi:10.1246/bcsj.53.478 |
| 11. | Enders, D.; Breuer, K.; Teles, J. H. Helv. Chim. Acta 1996, 79, 1217–1221. doi:10.1002/hlca.19960790427 |
| 12. | Knight, R. L.; Leeper, F. J. J. Chem. Soc., Perkin Trans. 1 1998, 1891–1894. doi:10.1039/A803635G |
| 13. | Enders, D.; Kallfass, U. Angew. Chem., Int. Ed. 2002, 41, 1743–1745. doi:10.1002/1521-3773(20020517)41:10<1743::AID-ANIE1743>3.0.CO;2-Q |
| 14. | Enders, D.; Niemeier, O. Angew. Chem., Int. Ed. 2006, 118, 1491–1495. doi:10.1002/anie.200503885 |
| 15. | Takikawa, H.; Hachisu, Y.; Bode, J. W.; Suzuki, K. Angew. Chem., Int. Ed. 2006, 118, 3572–3574. doi:10.1002/anie.200600268 |
| 16. | Enders, D.; Henseler, A. Adv. Synth. Catal. 2009, 351, 1749–1752. doi:10.1002/adsc.200900247 |
| 17. | Enders, D.; Grossmann, A.; Fronert, J.; Raabe, G. Chem. Commun. 2010, 46, 6282–6284. doi:10.1039/c0cc02013c |
| 28. | Bergmann, E. D.; Pelchowicz, Z. J. Org. Chem. 1954, 19, 1387–1390. doi:10.1021/jo01373a024 |
| 29. | Weitzberg, M.; Abu-Shakra, E.; Azeb, A.; Aizenshtat, Z.; Blum, J. J. Org. Chem. 1987, 52, 529–536. doi:10.1021/jo00380a010 |
| 30. | Tsantrizos, Y. S.; Folkins, P. L.; Britten, J. F.; Harpp, D. N.; Ogilvie, K. K. Can. J. Chem. 1992, 70, 158–164. doi:10.1139/v92-026 |
| 2. | Linghu, X.; Johnson, J. S. Angew. Chem. 2003, 115, 2638–2640. doi:10.1002/ange.200250554 |
| 3. | Tarr, J. C.; Johnson, J. S. Org. Lett. 2009, 11, 3870–3873. doi:10.1021/ol901314w |
| 4. | Bausch, C. C.; Johnson, J. S. Adv. Synth. Catal. 2005, 347, 1207–1211. doi:10.1002/adsc.200505097 |
| 5. | Demir, A. S.; Reis, Ö.; İğdir, A. Ç.; Esiringü, İ.; Eymur, S. J. Org. Chem. 2005, 70, 10584–10587. doi:10.1021/jo051811u |
| 31. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7 |
| 32. | Alpagut, Y.; Goldfuss, B.; Neudörfel, J.-M. Beilstein J. Org. Chem. 2008, 4, No. 25. doi:10.3762/bjoc.4.25 |
| 26. | Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 2, 256–263. doi:10.1002/ejoc.201001295 |
| 18. | Linghu, X.; Potnick, J. R.; Johnson, J. S. J. Am. Chem. Soc. 2004, 126, 3070–3071. doi:10.1021/ja0496468 |
| 26. | Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 2, 256–263. doi:10.1002/ejoc.201001295 |
| 26. | Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 2, 256–263. doi:10.1002/ejoc.201001295 |
| 21. | Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211–5218. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S |
| 22. | Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785–1792. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0 |
| 23. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273 |
| 24. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 25. | Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 27. | Nolan, B. G.; Tsujioka, S.; Strauss, S. H. J. Fluorine Chem. 2002, 118, 103–106. doi:10.1016/S0022-1139(02)00203-8 |
| 18. | Linghu, X.; Potnick, J. R.; Johnson, J. S. J. Am. Chem. Soc. 2004, 126, 3070–3071. doi:10.1021/ja0496468 |
| 26. | Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 2, 256–263. doi:10.1002/ejoc.201001295 |
| 19. | Goldfuss, B.; Schumacher, M. J. Mol. Model. 2006, 12, 591–595. doi:10.1007/s00894-005-0036-4 |
| 20. | Schumacher, M.; Goldfuss, B. Tetrahedron 2008, 64, 1648–1653. doi:10.1016/j.tet.2007.12.019 |
| 26. | Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 2, 256–263. doi:10.1002/ejoc.201001295 |
| 33. | Berger, S.; Braun, S.; Kalinowski, H.-O. NMR-Spektroskopie von Nichtmetallen Band 3. Thieme: Stuttgart, Germany, 1993; pp 121–122. |
© 2011 Gliga et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)