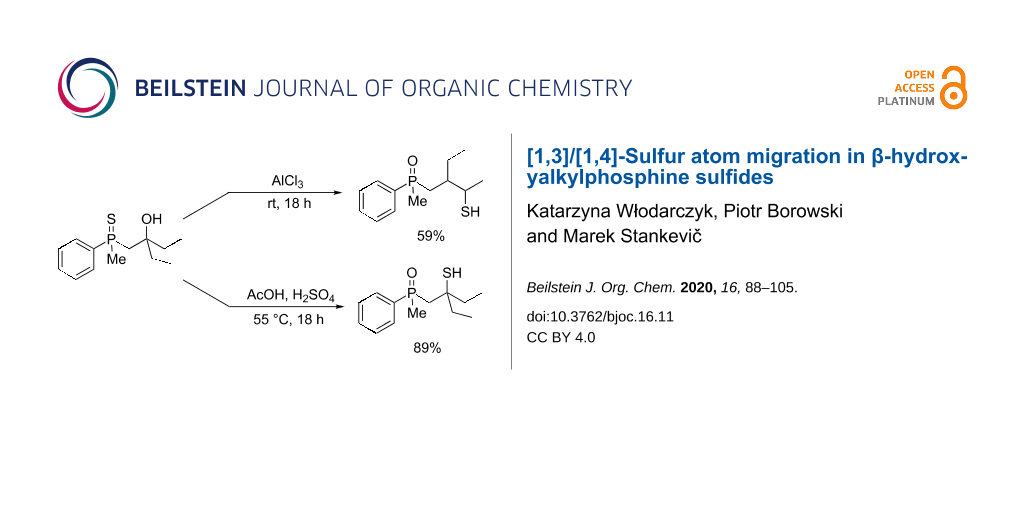
Abstract
β-Hydroxyalkylphosphine sulfides undergo [1,3]- or [1,4]-sulfur atom phosphorus-to-carbon migration in the presence of Lewis or Brønsted acids. The direction of sulfur atom migration depends on the type of acid used for the reaction. In the presence of a Brønsted acid, mainly [1,3]-rearrangement is observed, whereas a Lewis acid catalyzes the [1,4]-sulfur migration. To gain insight into the mechanism of these transformations, the stereochemistry of these rearrangements have been tested, along with the conduction of some control experiments and DFT calculations.

Graphical Abstract
Introduction
Among all organic reactions, rearrangements are an exciting class of transformations where unusual or even unexpected products can be obtained. Many rearrangements are of practical use in synthetic organic chemistry, including Beckmann [1-4], Claisen [5-8], pinacol [9-12], Wagner–Meerwein [13-16], Curtius [17-20], Hofmann [21-24], Overmann [25-28] rearrangements, and many others. In organophosphorus chemistry, rearrangements are less developed transformations, however, some examples can be found in the literature (Scheme 1). These include the famous Arbuzov rearrangement [29-32] as well as phospha-Fries ([1,3]-rearrangements) [33-37], phospha-Brook ([1,2]-rearrangement) [38-42], and [2,3]-sigmatropic rearrangements of propargyl and allylphosphinites [43-46].
![[1860-5397-16-11-i1]](/bjoc/content/inline/1860-5397-16-11-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Arbusov, phospha-Fries, and phospha-Brook rearrangements.
Scheme 1: Arbusov, phospha-Fries, and phospha-Brook rearrangements.
Other [1,2]-rearrangements include α,β-epoxyphosphonate opening [47], β-halo-α-hydroxyphosphonate rearrangement [48], phosphirene rearrangement [49], or the unusual phosphonate–phosphinate rearrangement of dimethyl phosphonates [50]. Other [1,3]-rearrangements of organophosphorus compounds include phosphoenolpyruvate formation from phosphonopyruvate [51], benzylphosphonium salt formation from the corresponding o-methylaryl-substituted precursors [52], or the formation of ketophosphonates from vinylphosphates [53]. [1,4]-Rearrangements include that of o-phosphorus-substituted benzyl carbanions [34], phosphorus group migration in O-phosphorylated 1,4-benzodiazepines [54], or phosphoryl group carbon-to-oxygen transfer [55]. The common feature of every rearrangement presented above is the cleavage of one single bond between phosphorus and either carbon or oxygen atom, while the multiple bond remains intact.
Herein, we present the results concerning an unusual transformation of β-hydroxyalkylphosphine sulfides, which undergo [1,3]- or [1,4]-rearrangement in the presence of an acid, yielding the corresponding β/γ-mercaptoalkylphosphine oxides. In this rearrangement, a sulfur atom transfers from phosphorus to carbon, whereas the phosphorus–carbon bonds remain intact. Depending on the type of acid, sulfur atom migration may occur at the β- or the γ-carbon atom.
Results and Discussion
In our previous papers [56,57], we described the intramolecular cationic cyclization of β-hydroxyalkylphosphine oxides (Scheme 2).
![[1860-5397-16-11-i2]](/bjoc/content/inline/1860-5397-16-11-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cyclization of 1a and 1b under acidic conditions.
Scheme 2: Cyclization of 1a and 1b under acidic conditions.
Depending on the structure, the formation of either the phosphaindane 3 or the benzophosphorinane 2 skeleton was observed. This method could also be performed in a stereoselective manner, given that corresponding chiral substrates were used for the cyclization. The chiral compounds suitable for cyclization could be obtained by desymmetrization of phosphine sulfides (Scheme 3) [58].
![[1860-5397-16-11-i3]](/bjoc/content/inline/1860-5397-16-11-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The synthesis of P-stereogenic β-hydroxyalkylphosphine sulfides.
Scheme 3: The synthesis of P-stereogenic β-hydroxyalkylphosphine sulfides.
In order to gain insight into the cationic cyclization of β-hydroxyalkylphosphine sulfides, a set of substrates 6–25 was prepared from dimethylphenylphosphine sulfide 4 (Table 1).
Table 1: Synthesis of substrates for this study.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-11-i15.svg?max-width=637&scale=1.0)
|
||||||
| entry | R | R’ | compound | yield, % | dr | |
| 1 | Me | H | 6 | 77 | 51:49 | |
| 2 | Et | H | 7 | 90 | 57:43 | |
| 3 | iPr | H | 8 | 75 | 55:45 | |
| 4 | t-Bu | H | 9 | 84 | 61.5:38.5 | |
| 5 | Ph | H | 10 | 71 | 60:40 | |
| 6 | cyclohexane | H | 11 | 89 | 57:43 | |
| 7 | Et | Me | 12 | 91 | 52:48 | |
| 8 | Ph | Me | 13 | 92 | 62.5:37.5 | |
| 9 | n-Bu | Me | 14 | 76 | 58:42 | |
| 10 | n-Pr | Me | 15 | 84 | 51:49 | |
| 11 | iPr | Me | 16 | 85 | 53:47 | |
| 12 | t-Bu | Me | 17 | 84 | 55:45 | |
| 13 | t-BuCH2 | Me | 18 | 86 | 53:47 | |
| 14 | Me | Me | 19 | 88 | ||
| 15 | Et | Et | 20 | 78 | ||
| 16 | iPr | iPr | 21 | 70 | ||
| 17 | –(CH2)4– | 22 | 72 | |||
| 18 | –(CH2)5– | 23 | 88 | |||
| 19 | –(CH2)6– | 24 | 88 | |||
| 20 | n-Bu | n-Bu | 25 | 78 | ||
All compounds were obtained in good yields, both from aldehydes and ketones. For aldehydes and unsymmetrically substituted ketones, the formation of the products as diastereomeric mixtures could be observed. Unfortunately, the diastereomeric excess of the reaction was low in each case.
In order to analyze the reactivity of β-hydroxyalkylphosphine sulfides under acidic conditions, the cyclization of 8 and 19 was attempted (Scheme 4).
![[1860-5397-16-11-i4]](/bjoc/content/inline/1860-5397-16-11-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cyclization of 8 and 19 in the presence of H3PO4.
Scheme 4: Cyclization of 8 and 19 in the presence of H3PO4.
Unexpectedly, when the reaction was performed under conditions developed originally for β-hydroxyalkylphosphine oxides [56,57], this led to the formation of cyclic phosphine oxides 3 and 26 rather than phosphine sulfides. It may be assumed that hydrolysis of the P=S bond occurred under these reaction conditions and, probably, racemization of the phosphorus center in the case of nonracemic β-hydroxyalkylphosphine sulfides. Indeed, the attempted cyclization of (SP)-19 under standard reaction conditions led to the formation of the completely racemic phosphine oxide 3 (Scheme 5).
![[1860-5397-16-11-i5]](/bjoc/content/inline/1860-5397-16-11-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cyclization of (SP)-19 in the presence of H3PO4.
Scheme 5: Cyclization of (SP)-19 in the presence of H3PO4.
The above results clearly show that phosphoric acid was inappropriate for the cyclization of nonracemic β-hydroxyalkylphosphine sulfides, and therefore, an alternative methodology had to be developed for the synthesis of chiral organophosphorus compounds possessing either phosphaindane or benzophosphorinane skeletons. As such, it was decided to test the ability of Lewis acids to catalyze the cyclization of β-hydroxyalkylphosphine sulfides (Table 2).
Table 2: Reaction of β-hydroxyalkylphosphine sulfides with Lewis-acidic AlCl3.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-11-i16.svg?max-width=637&scale=1.0)
|
||
| entry | substrate | product (yield) |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-11-i17.svg?max-width=637&scale=1.0)
7 |
no reaction |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-11-i18.svg?max-width=637&scale=1.0)
8 |
no reaction |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-11-i19.svg?max-width=637&scale=1.0)
9 |
no reaction |
| 4 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-11-i20.svg?max-width=637&scale=1.0)
11 |
no reaction |
| 5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-16-11-i21.svg?max-width=637&scale=1.0)
12 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-16-11-i22.svg?max-width=637&scale=1.0)
29 (74%) |
| 6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-16-11-i23.svg?max-width=637&scale=1.0)
13 |
mixture |
| 7 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-16-11-i24.svg?max-width=637&scale=1.0)
14 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-16-11-i25.svg?max-width=637&scale=1.0)
30 (94%) |
| 8 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-16-11-i26.svg?max-width=637&scale=1.0)
15 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-16-11-i27.svg?max-width=637&scale=1.0)
31 (81%) |
| 9 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-16-11-i28.svg?max-width=637&scale=1.0)
16 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-16-11-i29.svg?max-width=637&scale=1.0)
32 (74%) |
| 10 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-16-11-i30.svg?max-width=637&scale=1.0)
17 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-16-11-i31.svg?max-width=637&scale=1.0)
33 (74%) |
| 11 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-16-11-i32.svg?max-width=637&scale=1.0)
18 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-16-11-i33.svg?max-width=637&scale=1.0)
34 (96%) |
| 12 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-16-11-i34.svg?max-width=637&scale=1.0)
19 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-16-11-i35.svg?max-width=637&scale=1.0)
|
| 13 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-16-11-i36.svg?max-width=637&scale=1.0)
20 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-16-11-i37.svg?max-width=637&scale=1.0)
37 (59%) |
| 14 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-16-11-i38.svg?max-width=637&scale=1.0)
21 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-16-11-i39.svg?max-width=637&scale=1.0)
38 (59%) |
| 15 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-16-11-i40.svg?max-width=637&scale=1.0)
22 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-16-11-i41.svg?max-width=637&scale=1.0)
39 (40%) |
| 16 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-16-11-i42.svg?max-width=637&scale=1.0)
23 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-16-11-i43.svg?max-width=637&scale=1.0)
40 (60%) |
| 17 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-16-11-i44.svg?max-width=637&scale=1.0)
24 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-16-11-i45.svg?max-width=637&scale=1.0)
41 (88%) |
| 18 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-16-11-i46.svg?max-width=637&scale=1.0)
25 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-16-11-i47.svg?max-width=637&scale=1.0)
42 (60%) |
aConversion based on 31P NMR spectroscopic analysis of the crude mixture.
As it turned out, the sulfides derived from aldehydes 7–9 and 11 failed to undergo a rearrangement (Table 2, entries 1–4). On the other hand, compounds derived from ketones 12, 14–17, and 20–25 underwent a ‘clean’ reaction under the conditions. However, instead of cyclization, a phosphorus-to-carbon sulfur migration took place, affording the corresponding mercaptoalkylphosphine oxides 29–33 and 37–42 with satisfying yields (Table 2, entries 5, 7–10, and 13–18). This sulfur atom phosphorus-to-carbon migration in phosphine sulfides has previously been mentioned only once in the literature [59]. Another example of such a migration has been reported by Creary and Innocencio for functionalized α-hydroxythiophosphonic acid diesters [60].
What is more interesting, the sulfur atom did not simply replace oxygen at the β-carbon atom: in every case, the mercapto group was placed at the γ-carbon atom, suggesting that the reaction followed a more complex mechanism. All reactions tested proceeded with the formation of an additional chirality center at the γ-carbon atom in the alkyl substituent, and therefore, almost all products were obtained as mixtures of epimers. Interestingly, compound 18 underwent isobutene elimination rather than rearrangement under the reaction conditions, whereas compound 19 led to the formation of a mixture of three products, with alkenylphosphine sulfide 35 being the major compound.
The structures were assigned based on NMR analysis as all compounds failed to give single crystals for X-ray analysis. The presence of an oxygen atom attached to the phosphorus atom in the products could be deduced based on 1H and 13C NMR analysis. In the 1H NMR spectra of phosphine sulfide 12, the chemical shifts of the signals belonging to the aromatic protons differed significantly more compared to the appropriate phosphine oxide. In 29 (a mixture of two diastereomers), the same chemical shifts were remarkably closer to each other (Figure 1).
![[1860-5397-16-11-1]](/bjoc/content/figures/1860-5397-16-11-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectra of compounds 12 and 29.
Figure 1: 1H NMR spectra of compounds 12 and 29.
In addition, signals of methyl groups attached to phosphorus could be found at 1.70 and 1.71 ppm, respectively, which was in accordance with the shifts of this group in other phosphine oxides [61-63]. Moreover, the two signals at 2.91–2.98 and 3.16–3.24 ppm, respectively, corresponded to a hydrogen atom bonded to a sulfur atom [64,65].
Even more conclusive was the analysis of the 13C NMR spectra of 12 and 29 (Figure 2).
![[1860-5397-16-11-2]](/bjoc/content/figures/1860-5397-16-11-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 13C NMR spectra of compounds 12 and 29.
Figure 2: 13C NMR spectra of compounds 12 and 29.
In the 13C NMR spectra of compound 12, the signal of the ipso-carbon atom could be found at 133.1 and 133.2 ppm, respectively (a mixture of two diastereomers), with 1J coupling constants of 76.9 and 78.1 Hz, respectively. In 29, the signal of the same carbon atom could be found at 133.6 and 134.2 ppm, respectively, with 1J coupling constants of 95.8 and 93.9 Hz, respectively, values typical for arylphosphine derivatives possessing a P=O bond [61,63,66]. Similarly, shifts and coupling constants of CH3 groups bound to phosphorus (17.2 ppm/70.3 Hz and 17.7 ppm/70.3 Hz) and α-CH2 groups (35.4 ppm/69.7 Hz and 36.0 ppm/69.7 Hz) indicated the presence of a P=O bond within the molecule. The biggest difference, however, was seen for the C–X bond. For 12, the signals of the tertiary carbinolic carbon atom were found at 73.1 ppm/4.3 Hz and 73.3 ppm/4.3 Hz, respectively. For 29, the same signals were found at 41.2 ppm/11.0 Hz and 41.6 ppm/10.7 Hz, respectively. The shifts indicated the presence of a C–S and not a C–O bond in the carbon skeleton [64,65], and the coupling constant value was typical for an γ-carbon atom present in a phosphine derivative [67-69].
In this regard, 31P NMR analysis provides very little information about the reaction mechanism. For substrates 6–25, the 31P NMR shifts were in the range of 32.66–39.33 ppm, which was the typical range for aryldialkylphosphine sulfides [70,71]. The signal of aryldialkylphosphine oxides are usually found around 30–50 ppm [61,72,73], and hence the overlapping signals make this technique inappropriate for the analysis of the reaction products.
The outcome of the above-discussed reaction may suggest that the substrate underwent water elimination followed by intramolecular carbon–sulfur bond formation, and that the formed cationic intermediates underwent hydrolysis during aqueous workup, leading to the observed products. It was assumed that a similar sulfur atom migration should occur in the presence of Brønsted acid under dry conditions. Therefore, a set of phosphine sulfides prepared above was subjected to the reaction using a H2SO4/AcOH mixture (Table 3).
Table 3: Reaction of β-hydroxyalkylphosphine sulfides with Brønsted-acidic H2SO4.
![[Graphic 34]](/bjoc/content/inline/1860-5397-16-11-i48.svg?max-width=637&scale=1.0)
|
||
| entry | substrate | product (yield) |
| 1 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-16-11-i49.svg?max-width=637&scale=1.0)
6 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-16-11-i50.svg?max-width=637&scale=1.0)
43 (83%) |
| 2 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-16-11-i51.svg?max-width=637&scale=1.0)
7 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-16-11-i52.svg?max-width=637&scale=1.0)
44 (95%) |
| 3 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-16-11-i53.svg?max-width=637&scale=1.0)
8 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-16-11-i54.svg?max-width=637&scale=1.0)
|
| 4 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-16-11-i55.svg?max-width=637&scale=1.0)
9 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-16-11-i56.svg?max-width=637&scale=1.0)
47 (34%)a (25%) |
| 5 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-16-11-i57.svg?max-width=637&scale=1.0)
11 |
![[Graphic 44]](/bjoc/content/inline/1860-5397-16-11-i58.svg?max-width=637&scale=1.0)
|
| 6 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-16-11-i59.svg?max-width=637&scale=1.0)
12 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-16-11-i60.svg?max-width=637&scale=1.0)
50 (87%) |
| 7 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-16-11-i61.svg?max-width=637&scale=1.0)
13 |
mixture of products |
| 8 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-16-11-i62.svg?max-width=637&scale=1.0)
14 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-16-11-i63.svg?max-width=637&scale=1.0)
51 (88%) |
| 9 |
![[Graphic 50]](/bjoc/content/inline/1860-5397-16-11-i64.svg?max-width=637&scale=1.0)
15 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-16-11-i65.svg?max-width=637&scale=1.0)
52 (95%) |
| 10 |
![[Graphic 52]](/bjoc/content/inline/1860-5397-16-11-i66.svg?max-width=637&scale=1.0)
16 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-16-11-i67.svg?max-width=637&scale=1.0)
32 (79%) |
| 11 |
![[Graphic 54]](/bjoc/content/inline/1860-5397-16-11-i68.svg?max-width=637&scale=1.0)
18 |
mixture of products |
| 12 |
![[Graphic 55]](/bjoc/content/inline/1860-5397-16-11-i69.svg?max-width=637&scale=1.0)
19 |
![[Graphic 56]](/bjoc/content/inline/1860-5397-16-11-i70.svg?max-width=637&scale=1.0)
53 (69%) |
| 13 |
![[Graphic 57]](/bjoc/content/inline/1860-5397-16-11-i71.svg?max-width=637&scale=1.0)
20 |
![[Graphic 58]](/bjoc/content/inline/1860-5397-16-11-i72.svg?max-width=637&scale=1.0)
54 (89%) |
| 14 |
![[Graphic 59]](/bjoc/content/inline/1860-5397-16-11-i73.svg?max-width=637&scale=1.0)
21 |
![[Graphic 60]](/bjoc/content/inline/1860-5397-16-11-i74.svg?max-width=637&scale=1.0)
38 (91%) |
| 15 |
![[Graphic 61]](/bjoc/content/inline/1860-5397-16-11-i75.svg?max-width=637&scale=1.0)
22 |
![[Graphic 62]](/bjoc/content/inline/1860-5397-16-11-i76.svg?max-width=637&scale=1.0)
55 (26%) |
| 16 |
![[Graphic 63]](/bjoc/content/inline/1860-5397-16-11-i77.svg?max-width=637&scale=1.0)
23 |
![[Graphic 64]](/bjoc/content/inline/1860-5397-16-11-i78.svg?max-width=637&scale=1.0)
56 (96%) |
| 17 |
![[Graphic 65]](/bjoc/content/inline/1860-5397-16-11-i79.svg?max-width=637&scale=1.0)
24 |
![[Graphic 66]](/bjoc/content/inline/1860-5397-16-11-i80.svg?max-width=637&scale=1.0)
57 (27%) |
| 18 |
![[Graphic 67]](/bjoc/content/inline/1860-5397-16-11-i81.svg?max-width=637&scale=1.0)
25 |
![[Graphic 68]](/bjoc/content/inline/1860-5397-16-11-i82.svg?max-width=637&scale=1.0)
58 (37%) |
aYield of the product as isolated as a mixture with substrate. bt = 96 h.
It was found that the outcome of each reaction depended on the structure of the substrate. β-Hydroxyalkylphosphine sulfides 6–9 and 11, derived from aldehydes, underwent estrification reactions, yielding the corresponding acetates 43–45, 47, and 48 (Table 3, entries 1–5). On the other hand, most of the substrates derived from ketones 12–16 and 18–25 underwent intramolecular sulfur migration with the formation of mainly β-mercaptoalkylphosphine oxides in very good yields (Table 3, entries 6, 8–10, and 12–18). Only in the case of compounds 16 and 21, the corresponding γ-mercaptoalkylphosphine oxides 32 and 38 could be isolated as the main products, and both 13 and 18 afforded product mixtures (Table 3, entries 7 and 11). Generally, the rearrangement proceeded efficiently and most of the products were isolated in good yields except from compounds possessing cyclopentyl and cycloheptyl fragments, which afforded the β-mercaptophosphine oxides in low yields.
In order to gain insight into the reaction mechanism when using Brønsted acid, it was decided to perform the reactions at different temperatures and to monitor the reaction mixture at different times (Table 4).
Table 4: Control experiments under Brønsted acid catalysis.
![[Graphic 69]](/bjoc/content/inline/1860-5397-16-11-i83.svg?max-width=637&scale=1.0)
|
||||
| entry | T, °C | T, h | yield of 54a, % | yield of 59a, % |
| 1 | 55 | 1 | 93 | 7 |
| 2 | 55 | 2 | 94 | 6 |
| 3 | 55 | 24 | 100 | 0 |
| 4 | 25 | 1 | 17 | 83 |
| 5 | 25 | 2 | 30 | 70 |
| 6 | 25 | 4 | 56 | 44 |
| 7 | 25 | 24 | 100 | 0 |
aConversion based on NMR analysis of the crude reaction mixture.
When the reaction was performed at 55 °C, the conversion of substrate 20 to the corresponding product 54 reached 93% after 1 hour (Table 4, entry 1). Apart from this product, the presence of alkenylphosphine sulfide 59 was detected in the reaction mixture. Prolonged heating led to the consumption of alkene 59 and its transformation to 54. For the reaction performed at 25 °C, the principle observations remained the same except that the reaction speed was remarkably lower. Nevertheless, these experiments allowed to conclude that the rearrangement proceeded via a two-step mechanism, with the first step being hydroxy group removal followed by intramolecular formation of a sulfur–carbon bond. The second step seemed to be slow, and the intermediate carbocation could be trapped in the form of an alkenylphosphine sulfide.
The comparison of the reactivity of the same substrate under two different reaction conditions clearly showed that distinct reaction mechanisms were responsible for the substrate’s transformation under Lewis and Brønsted acid catalysis, but both seemed to include the preliminary formation of alkenylphosphine sulfide. This was shown for the Brønsted acid-mediated reaction (Table 4) but remains to be proved for Lewis acid use. It was then decided to prepare substrates possessing double bonds at the β position (Scheme 6).
![[1860-5397-16-11-i6]](/bjoc/content/inline/1860-5397-16-11-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of the alkenylphosphine sulfides used in study.
Scheme 6: Synthesis of the alkenylphosphine sulfides used in study.
It appeared that the sulfides derived from aldehydes 8 and 9 failed to undergo transformation to the corresponding alkenylphosphine sulfides when treated with a MsCl/NEt3 mixture. Instead, the corresponding mesylates 60 and 61 were isolated as the sole products (Scheme 6). The compounds derived from ketones 12, 16, and 19 underwent elimination reactions to the β,γ-alkenylphosphine sulfides 62, 64, and 65 under these conditions. For sulfide 12, a mixture of two β-alkenylphosphine sulfides 62 and 63 (a mixture of E- and Z-isomer) was obtained under each reaction condition tested.
Once all the compounds had been prepared, it was decided to test the reactivity of mesylates 60 and 61 in the presence of AlCl3 (Scheme 7).
![[1860-5397-16-11-i7]](/bjoc/content/inline/1860-5397-16-11-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The reaction of mesylate compounds with Lewis-acidic AlCl3.
Scheme 7: The reaction of mesylate compounds with Lewis-acidic AlCl3.
The reaction of these compounds with aluminum chloride proceeded smoothly, leading to the corresponding γ-mercaptoalkylphosphine oxides in good yields. Surprisingly, the reactivity of the mesylates 60 and 61 appeared to be completely different from the parent β-hydroxyalkylphosphine sulfides 8 and 9, which failed to undergo a rearrangement (see Table 2, entries 2 and 3). The difference must therefore have arisen from the different C–O bond dissociation energies of the alcohols and mesylates.
Assuming that the Lewis acid coordinated to either hydroxy oxygen atom or to one of the oxygen atoms of the mesyl group, the activation energies for C–O bond dissociation are ca. 34.6 kcal/mol for the secondary alcohol 8, ca. 23.3 kcal/mol for the mesylate 60, and ca. 25.0 kcal/mol for the tertiary alcohol 20, as estimated by DFT calculations. The value for mesylate roughly fitted the range according to which the reaction may proceed at room temperature. For mesylate 61, with a tert-butyl group at the β-carbon atom, the initial dissociation of the C–O bond must have been followed by CH3 group migration before the observed sulfur transfer took place.
Analogous attempts were made for compounds 62–65 (Scheme 8).
![[1860-5397-16-11-i8]](/bjoc/content/inline/1860-5397-16-11-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: The reaction of alkenylphosphine sulfides with AlCl3.
Scheme 8: The reaction of alkenylphosphine sulfides with AlCl3.
A mixture of alkenylphosphine sulfides 62 and 63 afforded the same γ-mercaptoalkylphosphine oxide 29 in good yield in the presence of AlCl3. Similarly, sulfide 64 afforded the phosphine oxide 32, possessing a mercapto group at the γ-carbon atom, under the same reaction conditions. Finally, sulfide 65 underwent a transformation into oxide 53, possessing an SH group at the β-carbon atom, under the reaction conditions. The latter result was slightly different from the two previous ones, as here, the sulfur atom migrated to the β and not the γ-carbon atom. Moreover, the parent alcohol 19 underwent mainly a dehydration reaction and the formation of α,β-alkenylphosphine sulfide 35 in the presence of AlCl3.
All of these experiments suggested that the transformation of β-hydroxyalkylphosphine sulfides in the presence of Lewis acid most probably proceeded through the initial formation of an intermediate alkenylphosphine sulfide, which then underwent an intramolecular reaction leading to a formal [1,4]-rearrangement product. It seemed that the position of the double bond was crucial for the rearrangement, which had to be situated between the β- and the γ-carbon atom. In this position, there was no coupling with the thiophosphoryl group, and the double bond retained its nucleophilic character. This allowed the coordination of an AlCl3 molecule to the C=C bond, which then became susceptible to an intramolecular nucleophilic attack by a sulfur atom. For α,β-alkenylphosphine sulfide 35, the conjugation of the double bond with thiophosphoryl fragment made the activation by AlCl3 impossible, which prevented the rearrangement.
To gain detailed insights into the possible reaction mechanism, DFT calculations were performed on model β-hydroxyalkylphosphine sulfide 20, possessing two ethyl groups at the β-carbon atom. First, the Brønsted acid-catalyzed rearrangement was investigated by molecular modeling (Scheme 9).
![[1860-5397-16-11-i9]](/bjoc/content/inline/1860-5397-16-11-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Rearrangement of 20 in the presence of Brønsted acid. The calculated energies next to the arrows are reported in kcal/mol.
Scheme 9: Rearrangement of 20 in the presence of Brønsted acid. The calculated energies next to the arrows ar...
The first step of the rearrangement involved the protonation of 20, which afforded intermediate I, where the proton inserted between the sulfur and the oxygen atom. This intermediate appeared to be stable and failed to undergo further transformation, according to calculations. The reason of this behavior was most probably the cyclic structure of the intermediate. However, if the intermediate I can undergo a second protonation process, intermediate III forms through transition state II. Water elimination from III proceeded with slight increase in energy, affording intermediate IV, which may undergo two different transformations: The first transformation involves dissociation of a water molecule, which leads to the formation of the dication IX through transition state VIII. Taking into account that all energy barriers were well below 10 kcal/mol, it seemed that this transformation was reversible, and therefore the dication IV may have entered an alternative pathway, which involved the rotation of the molecule around the Cα–Cβ bond (activation barrier: +5.4 kcal/mol, stabilization: −3.4 kcal/mol), which was feasible under the reaction conditions. The formed dication V underwent an intramolecular nucleophilic substitution through transition state VI, which proceeded with a small activation barrier, leading to the cyclic intermediate VII and further to the rearranged product 54.
The tentative mechanism discussed above does not explain the formation of the γ-mercaptoalkylphosphine sulfides from β-hydroxyalkylphosphine sulfides 16 and 21 and, to some extent, 8 and 11. In these cases, DFT calculations allowed to propose a slightly different mechanism (see Supporting Information File 1). In this case, a water molecule, formed after protonation of the OH group, facilitated proton transfer from the γ- to β-carbon atom via a process similar to the elimination/electrophilic addition to alkenes pathway.
The computational results discussed above showed that the crucial point for sulfur atom migration was the formation of a tertiary carbocation at the β-carbon atom, which was quite straightforward under these acidic conditions. To understand the completely different selectivity in Lewis acid-catalyzed rearrangements, DFT was again applied to the reaction between 20 and AlCl3 (Scheme 10).
![[1860-5397-16-11-i10]](/bjoc/content/inline/1860-5397-16-11-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Rearrangement of 20 in the presence of Lewis acid. The calculated energies next to the arrows are reported in kcal/mol.
Scheme 10: Rearrangement of 20 in the presence of Lewis acid. The calculated energies next to the arrows are r...
Compound 20 underwent a complexation with an AlCl3 molecule, leading to intermediate I, and the overall transformation proceeded with remarkable stabilization. In the next step, dissociation of the C−O bond occurred through transition state II, finally leading to alkene III. The activation energy for the C−O bond cleavage in 20, a tertiary alcohol, was similar to the value calculated for the mesylate 60 (23.3 kcal/mol) and was lower than that for β-hydroxyalkylphosphine sulfide 8, formally a secondary alcohol (34.6 kcal/mol).
One could consider an alternative mechanism for the formation of III including the six-membered transition state VII. In this case, proton migration would occur from the γ-carbon atom to chloride, resulting in the formation of HCl, Al(OH)Cl2, and intermediate III. However, all attempts to monitor this particular transition state failed.
The formed intermediate III underwent a reaction with a second AlCl3 molecule, which associated with the double bond, yielding IV in a slightly exothermic process. The formed intermediate possessing an activated C=C bond underwent intramolecular carbon–sulfur bond formation through the transition state V. In this case, the reaction proceeded solely at the γ-carbon atom, and all attempts directed at the discovery of the transition state where the sulfur attack occurred at the β-carbon atom failed. It was possible that the electron density of the C=C double bond was slightly shifted towards the β-carbon atom due to possible interaction with the P=S fragment, and coordination of an AlCl3 molecule additionally activated the γ-carbon atom for a sulfur attack.
Regarding the correctness of the proposed mechanism, two issues associated with the reactivity of 17 and 19, respectively, in the presence of AlCl3 must be discussed. The first is the formation of the adduct with tert-butyl methyl ketone, and the second is the formation of the adduct with acetone. For 17, the formation of β,γ-alkenylphosphine sulfide was not possible, whereas the transformation of 19 afforded mainly the α,β-unsaturated compound 35. DFT analysis of the C–O bond cleavage in the adducts analogous to I revealed two different pathways. A complex of 19 with AlCl3 underwent simultaneous C–O bond cleavage and α-hydrogen (but not γ-hydrogen) atom abstraction, which led to 35, with activation barrier of +24.8 kcal/mol and a stabilization energy of −16.1 kcal/mol. Yet, at the moment, this preference remains to be further investigated.
On the other hand, the complex of 17 and AlCl3 underwent C–O bond cleavage and γ-hydrogen atom abstraction, with an activation barrier of +16.5 kcal/mol, which led to the corresponding alkene, with a stabilization energy of −13.5 kcal/mol. The loss of AlCl3/H2O afforded the β,γ-alkenylphosphine sulfide, which then underwent coordination of the second AlCl3 molecule, with an overall slight stabilization (+6.0/−8.5 kcal/mol). The adduct analogous to IV underwent methyl group migration to the β-carbon atom, along with the formation of a covalent Al–C bond. The last step required a remarkable activation energy (31.0 kcal/mol), which was followed by slight stabilization of the formed γ-carbanion (−6.1 kcal/mol). The latter should then undergo intramolecular C–S bond formation, followed by hydrolysis, finally leading to the rearranged product.
It was then decided to investigate the stereoselectivity of the acid-catalyzed rearrangement using enantiomerically enriched phosphine sulfides. Therefore, the chiral substrates (SP)-60, (SP)-65, (SP)-19, and (SP)-20 were prepared from dimethylphenylphosphine sulfide 4 using sparteine chemistry (Scheme 11).
![[1860-5397-16-11-i11]](/bjoc/content/inline/1860-5397-16-11-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: The synthesis of chiral substrates for rearrangement reactions.
Scheme 11: The synthesis of chiral substrates for rearrangement reactions.
First, the enantioenriched alcohol (SP)-17, mesylate (SP)-60, and alkenylphosphine sulfide (SP)-65 were subjected to the reaction with Lewis-acidic AlCl3 (Scheme 12).
![[1860-5397-16-11-i12]](/bjoc/content/inline/1860-5397-16-11-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: The reaction of (SP)-60 and (SP)-65 with AlCl3.
Scheme 12: The reaction of (SP)-60 and (SP)-65 with AlCl3.
Rearrangement of the tertiary alcohol (SP)-17 proceeded very efficiently, affording the corresponding product in high yield. Unfortunately, the stereoselectivity of the reaction appeared to be quite low due to the significant racemization of the phosphorus center under the applied conditions. On the other hand, the treatment of (SP)-60 with Lewis acid led to the formation of chiral γ-mercaptoalkylphosphine oxide 46 in good yield and with only slight decrease in enantiomeric excess. The transformation of alkenylphosphine sulfide (SP)-65 under similar reaction conditions led to the formation of β-mercaptoalkylphosphine oxide 53, albeit in low yield and with a complete lack of stereoselectivity.
The results presented above show that, at least for mesylates, the sulfur atom migration may have been a stereospecific process. What was more striking, the complete racemization of (SP)-65 and the extended racemization of (SP)-17 occurred under the reaction conditions. This suggested that the rearrangement may have proceeded either via two different pathways, or one of the reaction components may have influence the rate of racemization. In the case of (SP)-17, the postulated dissociation of AlCl3⋅OH may have generated traces of HCl, which may have react with the cyclic intermediate, leading to a pentavalent compound. The latter would have been very susceptible to Berry or turnstile pseudorotation. This, in turn, should have led to a remarkable deterioration of the enantiomeric excess of the product. The same might be postulated for the rearrangement of alkenylphosphine sulfide 65, which should form a pentavalent intermediate equally well.
In the next step, the enantiomerically enriched β-hydroxyalkylphosphine sulfides (SP)-19 and (SP)-20 were subjected to the rearrangement in the presence of Brønsted-acidic H2SO4 (Scheme 13).
![[1860-5397-16-11-i13]](/bjoc/content/inline/1860-5397-16-11-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Reaction of chiral β-hydroxyalkylphosphine sulfides with Brønsted acid.
Scheme 13: Reaction of chiral β-hydroxyalkylphosphine sulfides with Brønsted acid.
It was found that phosphine sulfide (SP)-17 underwent efficient rearrangement, affording a mixture of three compounds: β-mercaptoalkylphosphine oxide 66, its S-acylated derivative 67, and S-acylated γ-mercaptoalkylphosphine oxide 68. Compound 66 was obtained with complete stereoselectivity, whereas the enantiomeric excess of both 67 and 68 was higher, 78% ee and 76% ee, respectively. The phosphine sulfides (SP)-19 and (SP)-20 underwent rearrangement to the corresponding chiral β-mercaptoalkylphosphine oxides 53 and 54 in good yields and with complete stereoselectivity as compared to the rearrangement of (SP)-17. Based on this, it can be concluded that sulfur atom migration occurred via a highly stereospecific pathway through an intramolecular substitution mechanism.
Here, the stereochemical differences in the outcome of the rearrangement of (SP)-17 must be noticed. In the presence of Lewis-acidic AlCl3, the rearrangement led effectively to the corresponding γ-mercaptoalkylphosphine oxide 33, albeit with a remarkable deterioration of enantiomeric excess. The rearrangement in the presence of Brønsted-acidic H2SO4 afforded only minor amounts of S-acylated γ-mercaptoalkylphosphine oxide 68, but the enantiomeric excess was even higher than in the substrate. This suggested that the [1,3]-rearrangement was stereospecific in the presence of Brønsted acid, whereas the [1,4]-rearrangement proceeded with additional partial enrichment of one enantiomer. The latter may have occurred through the preferential acylation of one of two diastereoisomers under the reaction conditions where the second chirality center at the phosphorus atom influenced the ratio of the acylation reaction.
With chiral β-mercaptoalkylphosphine sulfides in hand, it was decided to check their ability to undergo cyclization in the presence of AlCl3 (Scheme 14).
![[1860-5397-16-11-i14]](/bjoc/content/inline/1860-5397-16-11-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Attempted cyclization of enantiomerically enriched 53 and 46.
Scheme 14: Attempted cyclization of enantiomerically enriched 53 and 46.
These results differed remarkably from the reactivity of the analogous hydroxyalkylphosphine sulfides, which exclusively underwent sulfur atom migration. The nature of this difference is currently a subject of further research in our laboratory. Nevertheless, the above results opened a new pathway for the synthesis of bicyclic organophosphorus compounds possessing a chirality center at phosphorus. Moreover, the formation of the bicyclic compounds proceeded under mild reaction conditions and with a high degree of stereoselectivity.
Conclusion
In conclusion, in the present paper, we describe the rearrangement of a series of β-hydroxyalkylphosphine sulfides. Both experimental and theoretical analysis of the reaction mechanism were performed. Depending on the structure of the initial alcohol, the acid used (Brønsted or Lewis), and the reaction conditions (temperature, time), the acid-promoted transformations of the alcohols may lead to two different products: β- or γ-mercaptoalkylphosphine oxides. The formation of γ-mercaptoalkylphosphine oxides proceeded with the formation of an additional chirality center at the γ-carbon atom. Attempted rearrangements of P-stereogenic β-hydroxy- and β-mesyloxyalkylphosphine sulfides in the presence of either Brønsted or Lewis acid led to the corresponding rearranged products with variable degree of stereoselectivity. It was found that the P-stereogenic β-/γ-mercaptoalkylphosphine oxides underwent intramolecular cyclization to the corresponding bicyclic phosphine oxides with high stereoselectivity and high yield under mild reaction conditions.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, a mechanism for the rearrangement of 16 in the presence of Brønsted acid assessed by computational methods, and coordinates of computed compounds. | ||
| Format: PDF | Size: 996.0 KB | Download |
| Supporting Information File 2: Copies of NMR spectra of all pure compounds and their mixtures. | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
Lindsay, A. C.; Sperry, J. Tetrahedron 2017, 73, 4355–4362. doi:10.1016/j.tet.2017.05.093
Return to citation in text: [1] -
Maronna, M. M.; Kruissink, E. C.; Parton, R. F.; Tinge, J. T.; Hoelderich, W. F. Catal. Commun. 2017, 90, 23–26. doi:10.1016/j.catcom.2016.11.014
Return to citation in text: [1] -
Naik, P. N.; Alkobati, N. A. H.; Kusurkar, R. S. ARKIVOC 2015, No. vii, 362–376. doi:10.3998/ark.5550190.p009.291
Return to citation in text: [1] -
Kotha, S.; Ravikumar, O.; Majhi, J. Beilstein J. Org. Chem. 2015, 11, 1503–1508. doi:10.3762/bjoc.11.163
Return to citation in text: [1] -
Zhang, B.; Wang, X.; Cheng, C.; Sun, D.; Li, C. Angew. Chem., Int. Ed. 2017, 56, 7484–7487. doi:10.1002/anie.201704086
Return to citation in text: [1] -
Shymanska, N. V.; Pierce, J. G. Org. Lett. 2017, 19, 2961–2964. doi:10.1021/acs.orglett.7b01185
Return to citation in text: [1] -
Chan, C.-K.; Chen, Y.-H.; Tsai, Y.-L.; Chang, M.-Y. J. Org. Chem. 2017, 82, 3317–3326. doi:10.1021/acs.joc.7b00108
Return to citation in text: [1] -
Ricard, S.; Sanapo, G. F.; Rahem, N.; Daoust, B. J. Org. Chem. 2016, 81, 5066–5073. doi:10.1021/acs.joc.6b00512
Return to citation in text: [1] -
Gómez-Martínez, M.; Baeza, A.; Alonso, D. A. ChemCatChem 2017, 9, 1032–1039. doi:10.1002/cctc.201601362
Return to citation in text: [1] -
Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2016, 55, 15411–15414. doi:10.1002/anie.201609911
Return to citation in text: [1] -
Rao, C. N.; Khan, F. A. Org. Biomol. Chem. 2015, 13, 2768–2775. doi:10.1039/c4ob02423k
Return to citation in text: [1] -
Rudolf, O.; Rouchal, M.; Lyčka, A.; Klásek, A. Helv. Chim. Acta 2013, 96, 1905–1917. doi:10.1002/hlca.201300074
Return to citation in text: [1] -
Korda, A.; Pakulski, Z.; Cmoch, P.; Gwardiak, K.; Karczewski, R. Tetrahedron 2017, 73, 1740–1744. doi:10.1016/j.tet.2017.02.024
Return to citation in text: [1] -
Nishiyama, Y.; Yokoshima, S.; Fukuyama, T. Org. Lett. 2016, 18, 2359–2362. doi:10.1021/acs.orglett.6b00789
Return to citation in text: [1] -
Shen, Z.; Pan, X.; Lai, Y.; Hu, J.; Wan, X.; Li, X.; Zhang, H.; Xie, W. Chem. Sci. 2015, 6, 6986–6990. doi:10.1039/c5sc02485d
Return to citation in text: [1] -
Fu, J.-G.; Ding, R.; Sun, B.-F.; Lin, G.-Q. Tetrahedron 2014, 70, 8374–8379. doi:10.1016/j.tet.2014.08.072
Return to citation in text: [1] -
Zhou, H.; Hong, J.; Huang, J.; Chen, Z. Asian J. Org. Chem. 2017, 6, 817–820. doi:10.1002/ajoc.201700248
Return to citation in text: [1] -
Milbeo, P.; Moulat, L.; Didierjean, C.; Aubert, E.; Martinez, J.; Calmès, M. J. Org. Chem. 2017, 82, 3144–3151. doi:10.1021/acs.joc.7b00122
Return to citation in text: [1] -
He, X.; Cao, C.; Liang, J.; Li, X.; Zhang, T.; Meng, F. Synlett 2017, 28, 386–390. doi:10.1055/s-0036-1588905
Return to citation in text: [1] -
Zhang, D.; Zheng, H.; Wang, X. Tetrahedron 2016, 72, 1941–1953. doi:10.1016/j.tet.2016.02.060
Return to citation in text: [1] -
Miyamoto, K.; Yamashita, J.; Narita, S.; Sakai, Y.; Hirano, K.; Saito, T.; Wang, C.; Ochiai, M.; Uchiyama, M. Chem. Commun. 2017, 53, 9781–9784. doi:10.1039/c7cc05160c
Return to citation in text: [1] -
Huang, H.; Yang, Q.; Zhang, Q.; Wu, J.; Liu, Y.; Song, C.; Chang, J. Adv. Synth. Catal. 2016, 358, 1130–1135. doi:10.1002/adsc.201501071
Return to citation in text: [1] -
Jevtić, I.; Došen-Mićović, L.; Ivanović, E. R.; Ivanović, M. D. Synthesis 2016, 48, 1550–1560. doi:10.1055/s-0035-1561405
Return to citation in text: [1] -
Yoshimura, A.; Middleton, K. R.; Luedtke, M. W.; Zhu, C.; Zhdankin, V. V. J. Org. Chem. 2012, 77, 11399–11404. doi:10.1021/jo302375m
Return to citation in text: [1] -
Hennum, M.; Odden, H. H.; Gundersen, L.-L. Eur. J. Org. Chem. 2017, 846–860. doi:10.1002/ejoc.201601446
Return to citation in text: [1] -
Sharif, S. A. I.; Calder, E. D. D.; Harkiss, A. H.; Maduro, M.; Sutherland, A. J. Org. Chem. 2016, 81, 9810–9819. doi:10.1021/acs.joc.6b01881
Return to citation in text: [1] -
Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2016, 57, 1994–1996. doi:10.1016/j.tetlet.2016.03.087
Return to citation in text: [1] -
Rajasekaran, P.; Ansari, A. A.; Vankar, Y. D. Eur. J. Org. Chem. 2015, 2902–2913. doi:10.1002/ejoc.201500129
Return to citation in text: [1] -
Jasiak, A.; Mielniczak, G.; Owsianik, K.; Koprowski, M.; Krasowska, D.; Drabowicz, J. J. Org. Chem. 2019, 84, 2619–2625. doi:10.1021/acs.joc.8b03053
Return to citation in text: [1] -
Piekutowska, M.; Pakulski, Z. Tetrahedron Lett. 2007, 48, 8482–8486. doi:10.1016/j.tetlet.2007.09.162
Return to citation in text: [1] -
Matveeva, E. V.; Odinets, I. L.; Kozlov, V. A.; Shaplov, A. S.; Mastryukova, T. A. Tetrahedron Lett. 2006, 47, 7645–7648. doi:10.1016/j.tetlet.2006.08.050
Return to citation in text: [1] -
Bhattacharya, A. K.; Thyagarajan, G. Chem. Rev. 1981, 81, 415–430. doi:10.1021/cr00044a004
Return to citation in text: [1] -
Korb, M.; Lehrich, S. W.; Lang, H. J. Org. Chem. 2017, 82, 3102–3124. doi:10.1021/acs.joc.7b00030
Return to citation in text: [1] -
Berchel, M.; Salaün, J.-Y.; Couthon-Gourvès, H.; Haelters, J.-P.; Jaffrès, P.-A. Dalton Trans. 2010, 39, 11314–11316. doi:10.1039/c0dt00880j
Return to citation in text: [1] [2] -
Jayasundera, K. P.; Watson, A. J.; Taylor, C. M. Tetrahedron Lett. 2005, 46, 4311–4313. doi:10.1016/j.tetlet.2005.04.104
Return to citation in text: [1] -
Taylor, C. M.; Watson, A. J. Curr. Org. Chem. 2004, 8, 623–636. doi:10.2174/1385272043370717
Return to citation in text: [1] -
Legrand, O.; Brunel, J. M.; Constantieux, T.; Buono, G. Chem. – Eur. J. 1998, 4, 1061–1067. doi:10.1002/(sici)1521-3765(19980615)4:6<1061::aid-chem1061>3.0.co;2-v
Return to citation in text: [1] -
Zhang, F.; Jiang, M.; Liu, J.-T. Tetrahedron Lett. 2017, 58, 1871–1874. doi:10.1016/j.tetlet.2017.04.002
Return to citation in text: [1] -
Kondoh, A.; Aoki, T.; Terada, M. Chem. – Eur. J. 2017, 23, 2769–2773. doi:10.1002/chem.201605673
Return to citation in text: [1] -
Pallikonda, G.; Santosh, R.; Ghosal, S.; Chakravarty, M. Tetrahedron Lett. 2015, 56, 3796–3798. doi:10.1016/j.tetlet.2015.04.073
Return to citation in text: [1] -
Hayashi, M.; Nakamura, S. Angew. Chem., Int. Ed. 2011, 50, 2249–2252. doi:10.1002/anie.201007568
Return to citation in text: [1] -
Hammerschmidt, F.; Schmidt, S. Eur. J. Org. Chem. 2000, 2239–2245. doi:10.1002/1099-0690(200006)2000:12<2239::aid-ejoc2239>3.0.co;2-v
Return to citation in text: [1] -
Hasanov, H. H.; Ivanov, I. K.; Christov, V. C. Heteroat. Chem. 2017, 28, e21357. doi:10.1002/hc.21357
Return to citation in text: [1] -
Gomes, F.; Fadel, A.; Rabasso, N. J. Org. Chem. 2012, 77, 5439–5444. doi:10.1021/jo300790m
Return to citation in text: [1] -
Himbert, G.; Ruppmich, M.; Knöringer, H. J. Chin. Chem. Soc. 2003, 50, 143–151. doi:10.1002/jccs.200300020
Return to citation in text: [1] -
Demay, S.; Lotz, M.; Polborn, K.; Knochel, P. Tetrahedron: Asymmetry 2001, 12, 909–914. doi:10.1016/s0957-4166(01)00123-9
Return to citation in text: [1] -
Kost, D.; Sprecher, M. S. Tetrahedron Lett. 1975, 16, 4483–4486. doi:10.1016/s0040-4039(00)91099-2
Return to citation in text: [1] -
Fischer, G. W.; Schneider, P. J. Prakt. Chem. 1977, 319, 399–407. doi:10.1002/prac.19773190308
Return to citation in text: [1] -
Haber, S.; Schmitz, M.; Bergsträßer, U.; Hoffmann, J.; Regitz, M. Chem. – Eur. J. 1999, 5, 1581–1589. doi:10.1002/(sici)1521-3765(19990503)5:5<1581::aid-chem1581>3.0.co;2-2
Return to citation in text: [1] -
Qian, R.; Roller, A.; Hammerschmidt, F. J. Org. Chem. 2015, 80, 1082–1091. doi:10.1021/jo502567j
Return to citation in text: [1] -
Bowman, E.; McQueney, M.; Barry, R. J.; Dunaway-Mariano, D. J. Am. Chem. Soc. 1988, 110, 5575–5576. doi:10.1021/ja00224a054
Return to citation in text: [1] -
Solomon, S. A.; Allen, L. K.; Dane, S. B. J.; Wright, D. S. Eur. J. Inorg. Chem. 2014, 1615–1619. doi:10.1002/ejic.201301203
Return to citation in text: [1] -
An, Y.-Z.; An, J. G.; Wiemer, D. F. J. Org. Chem. 1994, 59, 8197–8202. doi:10.1021/jo00105a042
Return to citation in text: [1] -
Gololobov, Y. G.; Krasnova, I. Y.; Barabanov, S. V.; Khrustalev, V. N.; Andronati, S. A.; Pavlovsky, V. I. Tetrahedron Lett. 2014, 55, 4879–4882. doi:10.1016/j.tetlet.2014.07.010
Return to citation in text: [1] -
Sokolsky, A.; Smith, A. B., III. Org. Lett. 2012, 14, 4470–4473. doi:10.1021/ol3019709
Return to citation in text: [1] -
Włodarczyk, K.; Stankevič, M. Tetrahedron 2016, 72, 5074–5090. doi:10.1016/j.tet.2016.06.070
Return to citation in text: [1] [2] -
Włodarczyk, K.; Borowski, P.; Drach, M.; Stankevič, M. Tetrahedron 2017, 73, 239–251. doi:10.1016/j.tet.2016.12.008
Return to citation in text: [1] [2] -
Muci, A. R.; Campos, K. R.; Evans, D. A. J. Am. Chem. Soc. 1995, 117, 9075–9076. doi:10.1021/ja00140a028
Return to citation in text: [1] -
Chen, C. H.; Brighty, K. E. Tetrahedron Lett. 1980, 21, 4421–4424. doi:10.1016/s0040-4039(00)92189-0
Return to citation in text: [1] -
Creary, X.; Inocencio, P. A. J. Am. Chem. Soc. 1986, 108, 5979–5983. doi:10.1021/ja00279a052
Return to citation in text: [1] -
Stankevič, M.; Pisklak, J.; Włodarczyk, K. Tetrahedron 2016, 72, 810–824. doi:10.1016/j.tet.2015.12.043
Return to citation in text: [1] [2] [3] -
Hockless, D. C. R.; McDonald, M. A.; Pabel, M.; Wild, S. B. J. Organomet. Chem. 1997, 529, 189–196. doi:10.1016/s0022-328x(96)06641-7
Return to citation in text: [1] -
Rajendran, K. V.; Gilheany, D. G. Chem. Commun. 2012, 48, 817–819. doi:10.1039/c1cc14856g
Return to citation in text: [1] [2] -
Simian, H.; Robert, F.; Blank, I. J. Agric. Food Chem. 2004, 52, 306–310. doi:10.1021/jf035008h
Return to citation in text: [1] [2] -
Jin, L.; Wang, J.; Dong, G. Angew. Chem., Int. Ed. 2018, 57, 12352–12355. doi:10.1002/anie.201807760
Return to citation in text: [1] [2] -
Copey, L.; Jean-Gérard, L.; Framery, E.; Pilet, G.; Robert, V.; Andrioletti, B. Chem. – Eur. J. 2015, 21, 9057–9061. doi:10.1002/chem.201501324
Return to citation in text: [1] -
Dong, J.; Liu, L.; Ji, X.; Shang, Q.; Liu, L.; Su, L.; Chen, B.; Kan, R.; Zhou, Y.; Yin, S.-F.; Han, L.-B. Org. Lett. 2019, 21, 3198–3203. doi:10.1021/acs.orglett.9b00922
Return to citation in text: [1] -
Hu, G.; Chen, W.; Fu, T.; Peng, Z.; Qiao, H.; Gao, Y.; Zhao, Y. Org. Lett. 2013, 15, 5362–5365. doi:10.1021/ol402672e
Return to citation in text: [1] -
Binyamin, I.; Meidan-Shani, S.; Ashkenazi, N. Beilstein J. Org. Chem. 2015, 11, 1332–1339. doi:10.3762/bjoc.11.143
Return to citation in text: [1] -
Woźnicki, P.; Korzeniowska, E.; Stankevič, M. J. Org. Chem. 2017, 82, 10271–10296. doi:10.1021/acs.joc.7b01767
Return to citation in text: [1] -
Bruzik, K. S.; Stec, W. J. J. Org. Chem. 1990, 55, 6131–6135. doi:10.1021/jo00312a018
Return to citation in text: [1] -
Chen, Q.; Yan, X.; Du, Z.; Zhang, K.; Wen, C. J. Org. Chem. 2016, 81, 276–281. doi:10.1021/acs.joc.5b02308
Return to citation in text: [1] -
Pietrusiewicz, K. M.; Zabłocka, M.; Monkiewicz, J. J. Org. Chem. 1984, 49, 1522–1526. doi:10.1021/jo00183a009
Return to citation in text: [1]
| 64. | Simian, H.; Robert, F.; Blank, I. J. Agric. Food Chem. 2004, 52, 306–310. doi:10.1021/jf035008h |
| 65. | Jin, L.; Wang, J.; Dong, G. Angew. Chem., Int. Ed. 2018, 57, 12352–12355. doi:10.1002/anie.201807760 |
| 61. | Stankevič, M.; Pisklak, J.; Włodarczyk, K. Tetrahedron 2016, 72, 810–824. doi:10.1016/j.tet.2015.12.043 |
| 63. | Rajendran, K. V.; Gilheany, D. G. Chem. Commun. 2012, 48, 817–819. doi:10.1039/c1cc14856g |
| 66. | Copey, L.; Jean-Gérard, L.; Framery, E.; Pilet, G.; Robert, V.; Andrioletti, B. Chem. – Eur. J. 2015, 21, 9057–9061. doi:10.1002/chem.201501324 |
| 64. | Simian, H.; Robert, F.; Blank, I. J. Agric. Food Chem. 2004, 52, 306–310. doi:10.1021/jf035008h |
| 65. | Jin, L.; Wang, J.; Dong, G. Angew. Chem., Int. Ed. 2018, 57, 12352–12355. doi:10.1002/anie.201807760 |
| 1. | Lindsay, A. C.; Sperry, J. Tetrahedron 2017, 73, 4355–4362. doi:10.1016/j.tet.2017.05.093 |
| 2. | Maronna, M. M.; Kruissink, E. C.; Parton, R. F.; Tinge, J. T.; Hoelderich, W. F. Catal. Commun. 2017, 90, 23–26. doi:10.1016/j.catcom.2016.11.014 |
| 3. | Naik, P. N.; Alkobati, N. A. H.; Kusurkar, R. S. ARKIVOC 2015, No. vii, 362–376. doi:10.3998/ark.5550190.p009.291 |
| 4. | Kotha, S.; Ravikumar, O.; Majhi, J. Beilstein J. Org. Chem. 2015, 11, 1503–1508. doi:10.3762/bjoc.11.163 |
| 17. | Zhou, H.; Hong, J.; Huang, J.; Chen, Z. Asian J. Org. Chem. 2017, 6, 817–820. doi:10.1002/ajoc.201700248 |
| 18. | Milbeo, P.; Moulat, L.; Didierjean, C.; Aubert, E.; Martinez, J.; Calmès, M. J. Org. Chem. 2017, 82, 3144–3151. doi:10.1021/acs.joc.7b00122 |
| 19. | He, X.; Cao, C.; Liang, J.; Li, X.; Zhang, T.; Meng, F. Synlett 2017, 28, 386–390. doi:10.1055/s-0036-1588905 |
| 20. | Zhang, D.; Zheng, H.; Wang, X. Tetrahedron 2016, 72, 1941–1953. doi:10.1016/j.tet.2016.02.060 |
| 50. | Qian, R.; Roller, A.; Hammerschmidt, F. J. Org. Chem. 2015, 80, 1082–1091. doi:10.1021/jo502567j |
| 13. | Korda, A.; Pakulski, Z.; Cmoch, P.; Gwardiak, K.; Karczewski, R. Tetrahedron 2017, 73, 1740–1744. doi:10.1016/j.tet.2017.02.024 |
| 14. | Nishiyama, Y.; Yokoshima, S.; Fukuyama, T. Org. Lett. 2016, 18, 2359–2362. doi:10.1021/acs.orglett.6b00789 |
| 15. | Shen, Z.; Pan, X.; Lai, Y.; Hu, J.; Wan, X.; Li, X.; Zhang, H.; Xie, W. Chem. Sci. 2015, 6, 6986–6990. doi:10.1039/c5sc02485d |
| 16. | Fu, J.-G.; Ding, R.; Sun, B.-F.; Lin, G.-Q. Tetrahedron 2014, 70, 8374–8379. doi:10.1016/j.tet.2014.08.072 |
| 51. | Bowman, E.; McQueney, M.; Barry, R. J.; Dunaway-Mariano, D. J. Am. Chem. Soc. 1988, 110, 5575–5576. doi:10.1021/ja00224a054 |
| 9. | Gómez-Martínez, M.; Baeza, A.; Alonso, D. A. ChemCatChem 2017, 9, 1032–1039. doi:10.1002/cctc.201601362 |
| 10. | Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2016, 55, 15411–15414. doi:10.1002/anie.201609911 |
| 11. | Rao, C. N.; Khan, F. A. Org. Biomol. Chem. 2015, 13, 2768–2775. doi:10.1039/c4ob02423k |
| 12. | Rudolf, O.; Rouchal, M.; Lyčka, A.; Klásek, A. Helv. Chim. Acta 2013, 96, 1905–1917. doi:10.1002/hlca.201300074 |
| 48. | Fischer, G. W.; Schneider, P. J. Prakt. Chem. 1977, 319, 399–407. doi:10.1002/prac.19773190308 |
| 5. | Zhang, B.; Wang, X.; Cheng, C.; Sun, D.; Li, C. Angew. Chem., Int. Ed. 2017, 56, 7484–7487. doi:10.1002/anie.201704086 |
| 6. | Shymanska, N. V.; Pierce, J. G. Org. Lett. 2017, 19, 2961–2964. doi:10.1021/acs.orglett.7b01185 |
| 7. | Chan, C.-K.; Chen, Y.-H.; Tsai, Y.-L.; Chang, M.-Y. J. Org. Chem. 2017, 82, 3317–3326. doi:10.1021/acs.joc.7b00108 |
| 8. | Ricard, S.; Sanapo, G. F.; Rahem, N.; Daoust, B. J. Org. Chem. 2016, 81, 5066–5073. doi:10.1021/acs.joc.6b00512 |
| 49. | Haber, S.; Schmitz, M.; Bergsträßer, U.; Hoffmann, J.; Regitz, M. Chem. – Eur. J. 1999, 5, 1581–1589. doi:10.1002/(sici)1521-3765(19990503)5:5<1581::aid-chem1581>3.0.co;2-2 |
| 33. | Korb, M.; Lehrich, S. W.; Lang, H. J. Org. Chem. 2017, 82, 3102–3124. doi:10.1021/acs.joc.7b00030 |
| 34. | Berchel, M.; Salaün, J.-Y.; Couthon-Gourvès, H.; Haelters, J.-P.; Jaffrès, P.-A. Dalton Trans. 2010, 39, 11314–11316. doi:10.1039/c0dt00880j |
| 35. | Jayasundera, K. P.; Watson, A. J.; Taylor, C. M. Tetrahedron Lett. 2005, 46, 4311–4313. doi:10.1016/j.tetlet.2005.04.104 |
| 36. | Taylor, C. M.; Watson, A. J. Curr. Org. Chem. 2004, 8, 623–636. doi:10.2174/1385272043370717 |
| 37. | Legrand, O.; Brunel, J. M.; Constantieux, T.; Buono, G. Chem. – Eur. J. 1998, 4, 1061–1067. doi:10.1002/(sici)1521-3765(19980615)4:6<1061::aid-chem1061>3.0.co;2-v |
| 43. | Hasanov, H. H.; Ivanov, I. K.; Christov, V. C. Heteroat. Chem. 2017, 28, e21357. doi:10.1002/hc.21357 |
| 44. | Gomes, F.; Fadel, A.; Rabasso, N. J. Org. Chem. 2012, 77, 5439–5444. doi:10.1021/jo300790m |
| 45. | Himbert, G.; Ruppmich, M.; Knöringer, H. J. Chin. Chem. Soc. 2003, 50, 143–151. doi:10.1002/jccs.200300020 |
| 46. | Demay, S.; Lotz, M.; Polborn, K.; Knochel, P. Tetrahedron: Asymmetry 2001, 12, 909–914. doi:10.1016/s0957-4166(01)00123-9 |
| 61. | Stankevič, M.; Pisklak, J.; Włodarczyk, K. Tetrahedron 2016, 72, 810–824. doi:10.1016/j.tet.2015.12.043 |
| 72. | Chen, Q.; Yan, X.; Du, Z.; Zhang, K.; Wen, C. J. Org. Chem. 2016, 81, 276–281. doi:10.1021/acs.joc.5b02308 |
| 73. | Pietrusiewicz, K. M.; Zabłocka, M.; Monkiewicz, J. J. Org. Chem. 1984, 49, 1522–1526. doi:10.1021/jo00183a009 |
| 29. | Jasiak, A.; Mielniczak, G.; Owsianik, K.; Koprowski, M.; Krasowska, D.; Drabowicz, J. J. Org. Chem. 2019, 84, 2619–2625. doi:10.1021/acs.joc.8b03053 |
| 30. | Piekutowska, M.; Pakulski, Z. Tetrahedron Lett. 2007, 48, 8482–8486. doi:10.1016/j.tetlet.2007.09.162 |
| 31. | Matveeva, E. V.; Odinets, I. L.; Kozlov, V. A.; Shaplov, A. S.; Mastryukova, T. A. Tetrahedron Lett. 2006, 47, 7645–7648. doi:10.1016/j.tetlet.2006.08.050 |
| 32. | Bhattacharya, A. K.; Thyagarajan, G. Chem. Rev. 1981, 81, 415–430. doi:10.1021/cr00044a004 |
| 47. | Kost, D.; Sprecher, M. S. Tetrahedron Lett. 1975, 16, 4483–4486. doi:10.1016/s0040-4039(00)91099-2 |
| 25. | Hennum, M.; Odden, H. H.; Gundersen, L.-L. Eur. J. Org. Chem. 2017, 846–860. doi:10.1002/ejoc.201601446 |
| 26. | Sharif, S. A. I.; Calder, E. D. D.; Harkiss, A. H.; Maduro, M.; Sutherland, A. J. Org. Chem. 2016, 81, 9810–9819. doi:10.1021/acs.joc.6b01881 |
| 27. | Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2016, 57, 1994–1996. doi:10.1016/j.tetlet.2016.03.087 |
| 28. | Rajasekaran, P.; Ansari, A. A.; Vankar, Y. D. Eur. J. Org. Chem. 2015, 2902–2913. doi:10.1002/ejoc.201500129 |
| 67. | Dong, J.; Liu, L.; Ji, X.; Shang, Q.; Liu, L.; Su, L.; Chen, B.; Kan, R.; Zhou, Y.; Yin, S.-F.; Han, L.-B. Org. Lett. 2019, 21, 3198–3203. doi:10.1021/acs.orglett.9b00922 |
| 68. | Hu, G.; Chen, W.; Fu, T.; Peng, Z.; Qiao, H.; Gao, Y.; Zhao, Y. Org. Lett. 2013, 15, 5362–5365. doi:10.1021/ol402672e |
| 69. | Binyamin, I.; Meidan-Shani, S.; Ashkenazi, N. Beilstein J. Org. Chem. 2015, 11, 1332–1339. doi:10.3762/bjoc.11.143 |
| 21. | Miyamoto, K.; Yamashita, J.; Narita, S.; Sakai, Y.; Hirano, K.; Saito, T.; Wang, C.; Ochiai, M.; Uchiyama, M. Chem. Commun. 2017, 53, 9781–9784. doi:10.1039/c7cc05160c |
| 22. | Huang, H.; Yang, Q.; Zhang, Q.; Wu, J.; Liu, Y.; Song, C.; Chang, J. Adv. Synth. Catal. 2016, 358, 1130–1135. doi:10.1002/adsc.201501071 |
| 23. | Jevtić, I.; Došen-Mićović, L.; Ivanović, E. R.; Ivanović, M. D. Synthesis 2016, 48, 1550–1560. doi:10.1055/s-0035-1561405 |
| 24. | Yoshimura, A.; Middleton, K. R.; Luedtke, M. W.; Zhu, C.; Zhdankin, V. V. J. Org. Chem. 2012, 77, 11399–11404. doi:10.1021/jo302375m |
| 38. | Zhang, F.; Jiang, M.; Liu, J.-T. Tetrahedron Lett. 2017, 58, 1871–1874. doi:10.1016/j.tetlet.2017.04.002 |
| 39. | Kondoh, A.; Aoki, T.; Terada, M. Chem. – Eur. J. 2017, 23, 2769–2773. doi:10.1002/chem.201605673 |
| 40. | Pallikonda, G.; Santosh, R.; Ghosal, S.; Chakravarty, M. Tetrahedron Lett. 2015, 56, 3796–3798. doi:10.1016/j.tetlet.2015.04.073 |
| 41. | Hayashi, M.; Nakamura, S. Angew. Chem., Int. Ed. 2011, 50, 2249–2252. doi:10.1002/anie.201007568 |
| 42. | Hammerschmidt, F.; Schmidt, S. Eur. J. Org. Chem. 2000, 2239–2245. doi:10.1002/1099-0690(200006)2000:12<2239::aid-ejoc2239>3.0.co;2-v |
| 70. | Woźnicki, P.; Korzeniowska, E.; Stankevič, M. J. Org. Chem. 2017, 82, 10271–10296. doi:10.1021/acs.joc.7b01767 |
| 71. | Bruzik, K. S.; Stec, W. J. J. Org. Chem. 1990, 55, 6131–6135. doi:10.1021/jo00312a018 |
| 34. | Berchel, M.; Salaün, J.-Y.; Couthon-Gourvès, H.; Haelters, J.-P.; Jaffrès, P.-A. Dalton Trans. 2010, 39, 11314–11316. doi:10.1039/c0dt00880j |
| 52. | Solomon, S. A.; Allen, L. K.; Dane, S. B. J.; Wright, D. S. Eur. J. Inorg. Chem. 2014, 1615–1619. doi:10.1002/ejic.201301203 |
| 53. | An, Y.-Z.; An, J. G.; Wiemer, D. F. J. Org. Chem. 1994, 59, 8197–8202. doi:10.1021/jo00105a042 |
| 60. | Creary, X.; Inocencio, P. A. J. Am. Chem. Soc. 1986, 108, 5979–5983. doi:10.1021/ja00279a052 |
| 61. | Stankevič, M.; Pisklak, J.; Włodarczyk, K. Tetrahedron 2016, 72, 810–824. doi:10.1016/j.tet.2015.12.043 |
| 62. | Hockless, D. C. R.; McDonald, M. A.; Pabel, M.; Wild, S. B. J. Organomet. Chem. 1997, 529, 189–196. doi:10.1016/s0022-328x(96)06641-7 |
| 63. | Rajendran, K. V.; Gilheany, D. G. Chem. Commun. 2012, 48, 817–819. doi:10.1039/c1cc14856g |
| 56. | Włodarczyk, K.; Stankevič, M. Tetrahedron 2016, 72, 5074–5090. doi:10.1016/j.tet.2016.06.070 |
| 57. | Włodarczyk, K.; Borowski, P.; Drach, M.; Stankevič, M. Tetrahedron 2017, 73, 239–251. doi:10.1016/j.tet.2016.12.008 |
| 59. | Chen, C. H.; Brighty, K. E. Tetrahedron Lett. 1980, 21, 4421–4424. doi:10.1016/s0040-4039(00)92189-0 |
| 56. | Włodarczyk, K.; Stankevič, M. Tetrahedron 2016, 72, 5074–5090. doi:10.1016/j.tet.2016.06.070 |
| 57. | Włodarczyk, K.; Borowski, P.; Drach, M.; Stankevič, M. Tetrahedron 2017, 73, 239–251. doi:10.1016/j.tet.2016.12.008 |
| 58. | Muci, A. R.; Campos, K. R.; Evans, D. A. J. Am. Chem. Soc. 1995, 117, 9075–9076. doi:10.1021/ja00140a028 |
| 54. | Gololobov, Y. G.; Krasnova, I. Y.; Barabanov, S. V.; Khrustalev, V. N.; Andronati, S. A.; Pavlovsky, V. I. Tetrahedron Lett. 2014, 55, 4879–4882. doi:10.1016/j.tetlet.2014.07.010 |
| 55. | Sokolsky, A.; Smith, A. B., III. Org. Lett. 2012, 14, 4470–4473. doi:10.1021/ol3019709 |
© 2020 Włodarczyk et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)